
Abstract
Background:
Plenty of evidence has suggested that autophagy plays a crucial role in the biological processes of cancers. This study aimed to screen autophagy-related genes (ARGs) and establish a novel a scoring system for colorectal cancer (CRC).
Methods:
Autophagy-related genes sequencing data and the corresponding clinical data of CRC in The Cancer Genome Atlas were used as training data set. The GSE39582 data set from the Gene Expression Omnibus was used as validation set. An autophagy-related signature was developed in training set using univariate Cox analysis followed by stepwise multivariate Cox analysis and assessed in the validation set. Then we analyzed the function and pathways of ARGs using Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Finally, a prognostic nomogram combining the autophagy-related risk score and clinicopathological characteristics was developed according to multivariate Cox analysis.
Results:
After univariate and multivariate analysis, 3 ARGs were used to construct autophagy-related signature. The KEGG pathway analyses showed several significantly enriched oncological signatures, such as p53 signaling pathway, apoptosis, human cytomegalovirus infection, platinum drug resistance, necroptosis, and ErbB signaling pathway. Patients were divided into high- and low-risk groups, and patients with high risk had significantly shorter overall survival (OS) than low-risk patients in both training set and validation set. Furthermore, the nomogram for predicting 3- and 5-year OS was established based on autophagy-based risk score and clinicopathologic factors. The area under the curve and calibration curves indicated that the nomogram showed well accuracy of prediction.
Conclusions:
Our proposed autophagy-based signature has important prognostic value and may provide a promising tool for the development of personalized therapy.
Introduction
Colorectal cancer (CRC) is one of the most frequently diagnosed malignancies worldwide. According to the 2018 Global Cancer Statistics, CRC is the third most common cancer worldwide and leading cause of cancer-related deaths. 1 Despite recent advances in curative surgery and adjuvant therapy, the mortality is still high due to its late detection and high recurrence rates, and the 5-year survival rate of patients remains poor. 2 -4 Therefore, it is urgently necessary to develop sensitive, cost-effective, and noninvasive methods to complement and improve current screening strategies for CRC diagnosis and prognosis.
Autophagy is a cell self-digestive process, which is initiated from the formation of autophagosome to deliver the protein and cell organelle to lysosome for degradation. This catabolic process is involved in a variety of autophagy-related genes (ARGs), which could influence the activation of autophagy. 5 Under some pathological conditions such as inflammation, neurodegeneration, aging, and tumor, autophagy can be stimulated to maintain cellular homeostasis. 6,7 However, the role of autophagy in tumorigenesis remains controversial, and mechanism is still rudimentary and inconclusive. Since autophagy has complex functions in cancer, it is urgent to further explore the potential biological processes of autophagy in tumorigenesis and development.
The most commonly used approach for predicting patient survival remains pathological staging according to the tumor–node–metastasis (TNM) classification system. 8 However, this staging system is not sufficient to assess prognosis and does not reflect the biological heterogeneity of cancer. It provides only limited information for the clinical prognostication since even patients within the same stage display a strong heterogeneity for prognosis and treatment response. Other factors such as age, performance status, and tumor location can also influence the patients’ survival. 9 -11 In addition, this system is not very useful in predicting individual patient outcomes. Therefore, this highlights the critical need to develop robust prognostic biomarkers that can offer a superior prognostic clinical usefulness. In recent years, nomograms have gradually gained popularity. They are valuable tools to use different clinical variables to determine a statistical prognostic model that generates a probability of clinical outcomes for a single patient. 12 Nomograms have been applied in various types of cancers. 13,14 For example, Zhao et al14 used 5 bone metastases-related genes to establish a prognostic nomogram, which can help in prediction of early bone metastases for patients with breast cancer. Similarly, Peak et al 15 developed a nomogram to predict lymph node metastasis based on 3 variables, including tumor grade, lymphovascular invasion, and clinical lymph node status. To our best knowledge, no prior prognostic models for CRC were established based on a combination of ARGs and clinicopathological features. Therefore, the aim of the present study was to identify CRC-related autophagy genes and build a novel prognostic signature through a comprehensive analysis of TCGA sequencing data.
Materials and Methods
Data Acquisition
The autophagy gene list was downloaded from the Human Autophagy Database (HADb, http://autophagy.lu/clustering/index.html). The data of messenger RNA (mRNA)-sequencing and the corresponding clinical data in patients with CRC were collected from The Cancer Genome Atlas (TCGA) database. To validate the predictive ability of autophagy-related risk score, we obtained the GSE39582 data set from the Gene Expression Omnibus (GEO) database. Overall, 499 samples from the TCGA database were used as training set, 556 samples from GSE39582 data set were used as validation set. Approval by ethics committee would not be necessary because all data had been collected from public availability of data in the HADb, GEO, and TCGA databases.
The Differentially Expressed ARGs Screening
All genes were downloaded in FPKM format and the genes that met the cutoff criteria of P < .05 and fold change (FC) > 2.0 were screened out as differentially expressed ARGs. The R package limma was used to identify differentially expressed ARGs between normal samples and tumor samples.
Functional Enrichment Analysis
The screening of the functions of differentially expressed ARGs was analyzed by the Gene Ontology (GO), which could obtain the biological function of gene. In addition, the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was used to enrich the differential pathway by “clusterProfiler” package in R/Bioconductor. 16 The threshold for enrichment significance was P value <.05.
Signature Development and Validation
A univariate Cox proportional hazards model was used to screen out ARGs that are significantly correlated with the overall survival (OS) of patients with CRC. These ARGs with a P value <.05 by univariate analysis were subjected to a multivariate Cox regression analysis with stepwise selection of variables based on the Akaike information criterion for identifying optimal prognostic ARGs. A autophagy-related signature was constructed based on a linear combination of the relative expression level of genes multiplied by regression coefficients (β), which represented the relative weight of genes in the multiple Cox regression analysis. The risk score = (βmRNA1 × expression level of mRNA1) + (βmRNA2 × expression level of mRNA2) + (βmRNA3 × expression level of mRNA3) +…+ (βmRNAn × expression level of mRNAn). Patients were divided into high-risk group and low-risk group by the median risk score as the cutoff value. Kaplan-Meier curves were plotted and survival differences between high-risk and low-risk group were compared by the log-rank test. The concordance index (C-index) was used to assess the prediction accuracy of the risk score model. In addition, GSE14520 data set was downloaded from the GEO database as a validation set. The risk score for each patient was calculated with the same formula as training set. The Kaplan-Meier curve was used to assess the predictive ability of the autophagy-related signature.
Construction and Validation of Nomogram
To explore whether the autophagy-related signature could be independent of other clinicopathological parameters (including age, gender, tumor location, prior malignancy, pretreatment carcinoembryonic antigen (CEA), Kirsten rat sarcoma (KRAS) mutation, lymphatic invasion, tumor stage, primary therapy outcome, and radiation therapy), Cox proportional hazard regression was performed. Variables in the univariate analysis with P values <.05 were selected for multivariate analysis. A nomogram for predicting the 3-year and 5-year OS was established based on the result of multivariate Cox regression analysis. The internal validation of nomogram was performed by discrimination and calibration. The discrimination was estimated by the area under the curve (AUC) of receiver operating characteristic (ROC) curve. 17 The AUC ranges from 0.5 to 1.0, with 0.5 indicates the outcomes is totally random and 1.0 indicates the perfect discrimination. The calibration was assessed by calibration curves, which shows how close the nomogram estimated risk was to the observed risk.
Statistical Analyses
Quantitative variables were compared using the t test or Mann-Whitney U test; the Pearson χ2 test or Fisher exact test was used to analyze categorical variables. Survival curves were created by the Kaplan-Meier method and compared using the log-rank test. The Cox regression coefficients were used to establish a risk score signature. The nomogram was built on the basis of multivariate analysis results by the package of “rms” in R. The prediction ability of nomogram was assessed by area under the ROC curve in the package of “survivalROC” in R. Calibration plots were used to compare the observed and predicted probabilities for the nomogram. All the statistical analyses were performed by SPSS 23.0 (Chicago, Illinois), along with version 3.5.3 of R software. Data were considered to be statistically significant when P value was less than .05.
Results
Identification of Differentially Expressed ARGs
The overall process of this study is described as Figure 1. In this work, we collected an mRNA expression data from TCGA data set, and it contained 571 CRC samples and 44 normal samples. A total of 231 ARGs were obtained from the HADb. Based on the cutoff criteria (P value <.05 and FC > 2.0), we identified 36 differentially expressed ARGs, including 16 upregulated and 20 downregulated genes. The expression of 36 differentially expressed ARGs between CRC and normal tissues was visualized by scatter plots (Figure 2). The clinicopathologic characteristics of patients with CRC are summarized in Table 1.
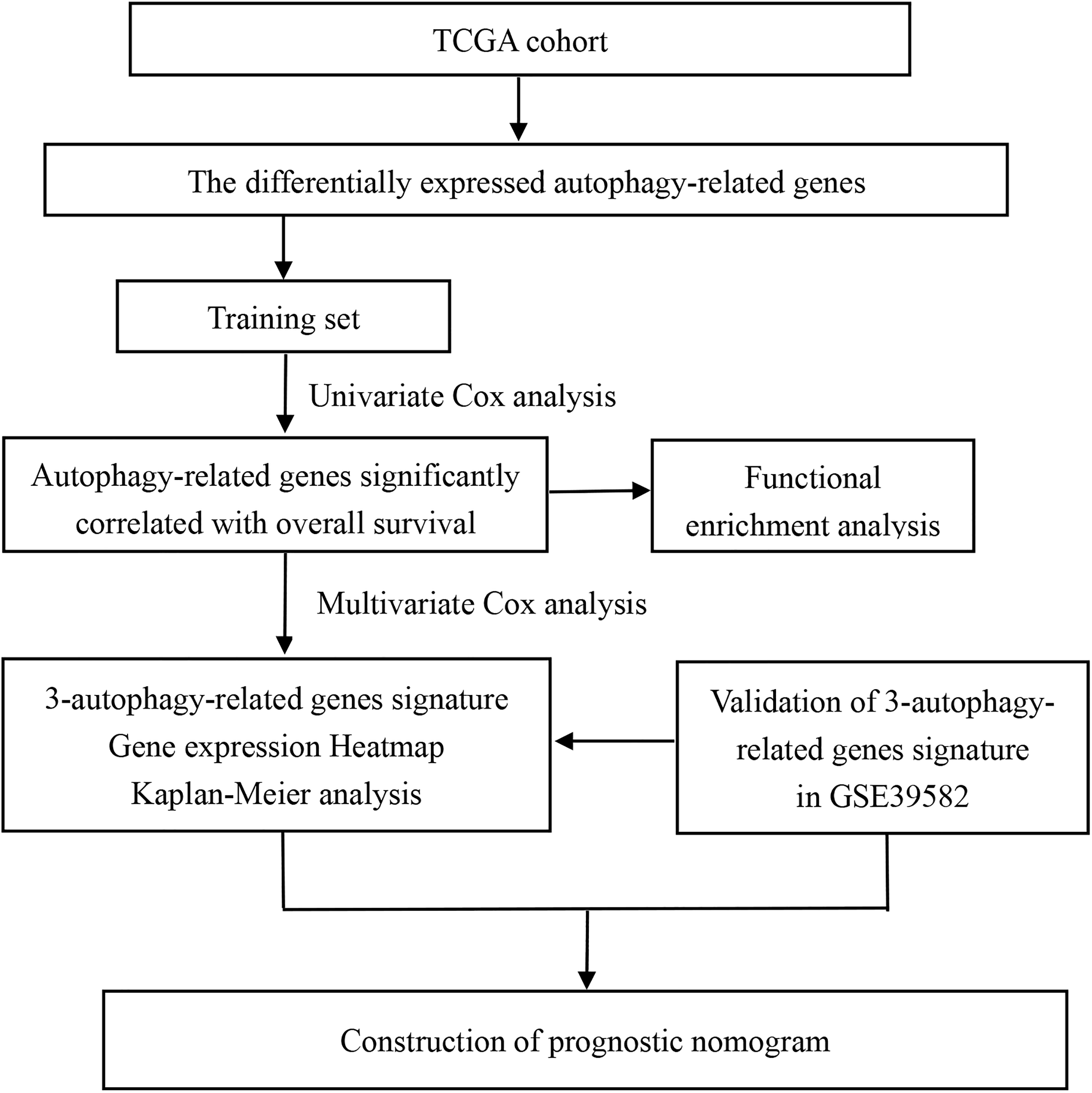
The flowchart identifying autophagy-related prognostic signature.

The expression of 36 differentially expressed autophagy-related genes between colorectal cancer and normal tissues.
Clinicopathologic Characteristics of Patients With CRC in TCGA and GEO Set.a

Abbreviations: CEA, carcinoembryonic antigen; CRC, colorectal cancer; GEO, Gene Expression Omnibus; KRAS, Kirsten rat sarcoma; SD, standard deviation; TCGA, The Cancer Genome Atlas.
a Other: Partial response, progressive disease, and stable disease.
Functional Enrichment Analysis
Thirty-six differentially expressed ARGs were annotated according to the GO database. We enriched the functions of these mRNA (P < .05). As shown in Figure 3A, autophagy and process utilizing autophagic mechanism were significantly enriched for biological processes. While for cellular component was autophagosome, for molecular functions were ubiquitin protein ligase binding and ubiquitin-like protein ligase binding. Furthermore, we analyzed KEGG pathways of these ARGs. We found certain KEGG pathways positively associated with ARGs such as p53 signaling pathway, apoptosis, human cytomegalovirus infection (HCMV), platinum drug resistance, necroptosis, and ErbB signaling pathway (Figure 3B).
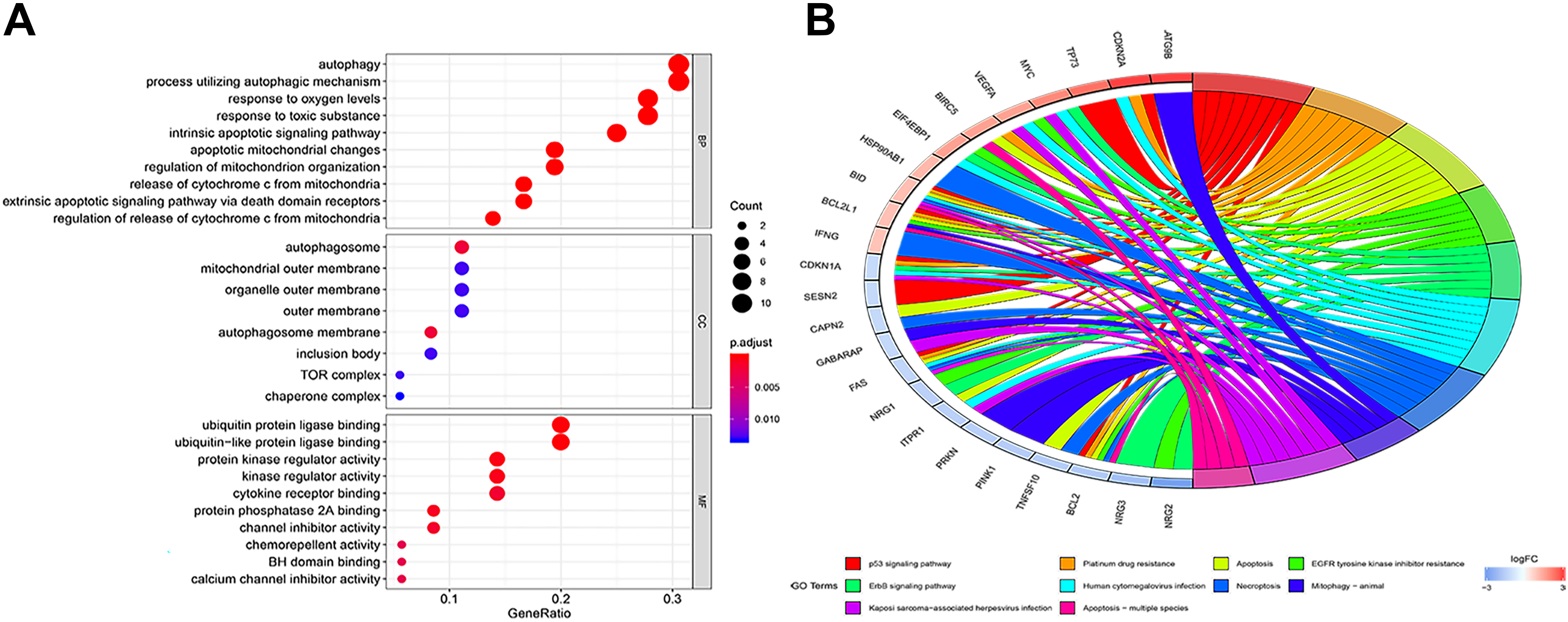
Enrichment of top 10 Gene Ontology terms (A) and Kyoto Encyclopedia of Genes and Genomes pathways (B) of differentially expressed autophagy-related genes. The node color changes gradually from red to blue in ascending order according to the adjust P values. The size of the node represents the number of counts.
Establishment of Autophagy-Related Signature in Training Set
To identify the OS-related ARGs, 36 differentially expressed ARGs were initially subjected to univariate Cox proportional hazards regression analysis in the training set. The result showed that 4 ARGs were significantly associated with the OS (P < .05). Then the 4 prognosis-related ARGs were subsequently selected for multivariate Cox proportional hazards regression. Finally, 3 ARGs (CDKN2A, NRG1, and VEGFA) were confirmed and used to establish an autophagy-related signature (Table 2). These include 2 potential risky genes and 1 potential protective gene. The risk score for OS was calculated as follows: risk score = 0.203082 × (expression value of CDKN2A) − 1.21191 × (expression value of NRG1) + 0.321454 × (expression value of VEGFA). We then calculated the risk score for each patient used this signature and used the median risk score as a cutoff value for classifying patient into a high-risk group (n = 249) and a low-risk group (n = 250), respectively. The heatmap, the risk score, and OS time of these 3 prognostic ARGs in training set are shown in Figure 4A. Kaplan-Meier curve and time-dependent ROC curves were used to evaluate the prognostic potential of the 3 autophagy-related signature. Kaplan-Meier curves showed that patients with high-risk group had a significantly lower OS than patients with the low-risk group (P < .001; Figure 4A). The C-index for OS was 0.784 for training set.
Three Prognostic ARGs Significantly Associated With OS in the TCGA Set.

Abbreviations: ARG, autophagy related gene; OS, overall survival; TCGA, The Cancer Genome Atlas.

The distribution of risk score, survival status, the heatmap, and Kaplan-Meier survival curves of 3-autophagy-related signature in training set (A) and in the validation set (B).
Validation of the Autophagy-Related Signature
To validate the predictive ability of autophagy-related signature, risk scores were calculated with the same formula for patient in GSE39582. Consistent with the results in the TCGA set, patients were categorized into high-risk group and low-risk group according to the median of the risk score. In the validation set, the distribution of survival status and risk scores of patients (Figure 4B) have a similar trend to that in the training set. Consistent with the results in the training set, survival analysis showed a significantly lower OS (P < .001) in the high-risk group (Figure 4B). The C-index for OS was 0.735 for validation set. Taking together, the autophagy-related signature was capable of predicting OS in CRC.
Construction and Validation of Nomogram
To integrate all independent risk factors for OS for the construction of prognostic nomogram, risk score and several clinicopathological factors were subjected to univariate and multivariate cox proportional hazards regression analyses. Univariate analysis demonstrated that age, pretreatment CEA, KRAS mutation, lymphatic invasion, stage, primary therapy outcome, radiation therapy, and risk score were all significant prognostic factors and corresponded to those for OS in the training set. All the significantly different variables were included in the multivariate analyses (Table 3). Finally, age, stage, primary therapy outcome, and risk score were significant risk factors for the patients with CRC (P < .05). A nomogram was constructed by multivariate analyses. A weighted total score calculated from each variable was used to estimate the 3- and 5-year OS of patients with CRC (Figure 5). The predictive abilities of the nomogram were analyzed by the AUC values. The AUC value of the nomogram predicting the 3- and 5-year OS rates were 0.797 and 0.763, respectively (Figure 6). In addition, we compared the performance of the nomogram with that of the American Joint Committee on Cancer (AJCC) TNM classification. The predictive ability of our nomogram was significantly superior to that of the TNM staging systems in the training set (Figure 6). The calibration plots showed well correlation between observed OS and nomogram predicted OS (Figure 7).
Univariate and Multivariate Analyses of Overall Survival in TCGA Set.
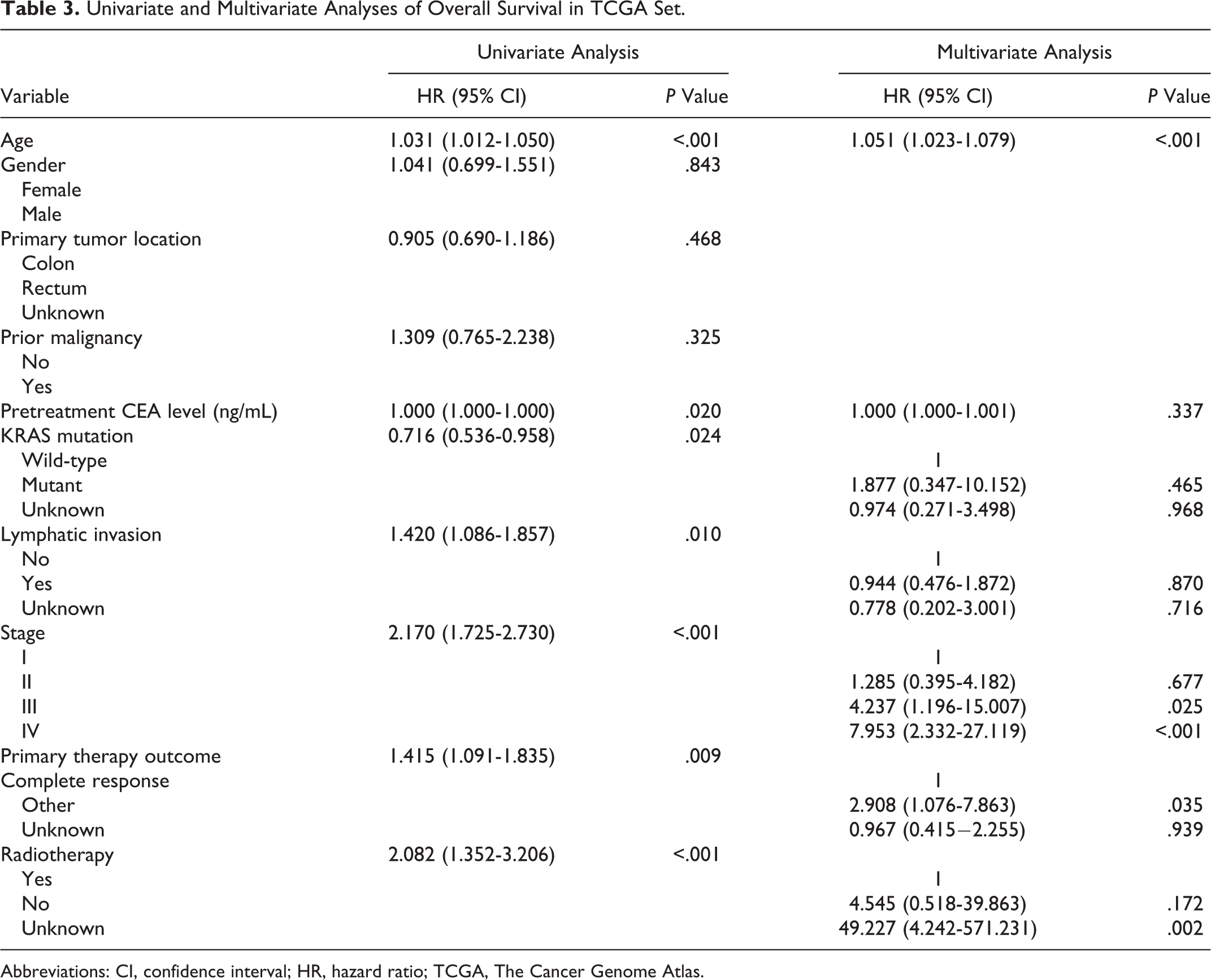
Abbreviations: CI, confidence interval; HR, hazard ratio; TCGA, The Cancer Genome Atlas.
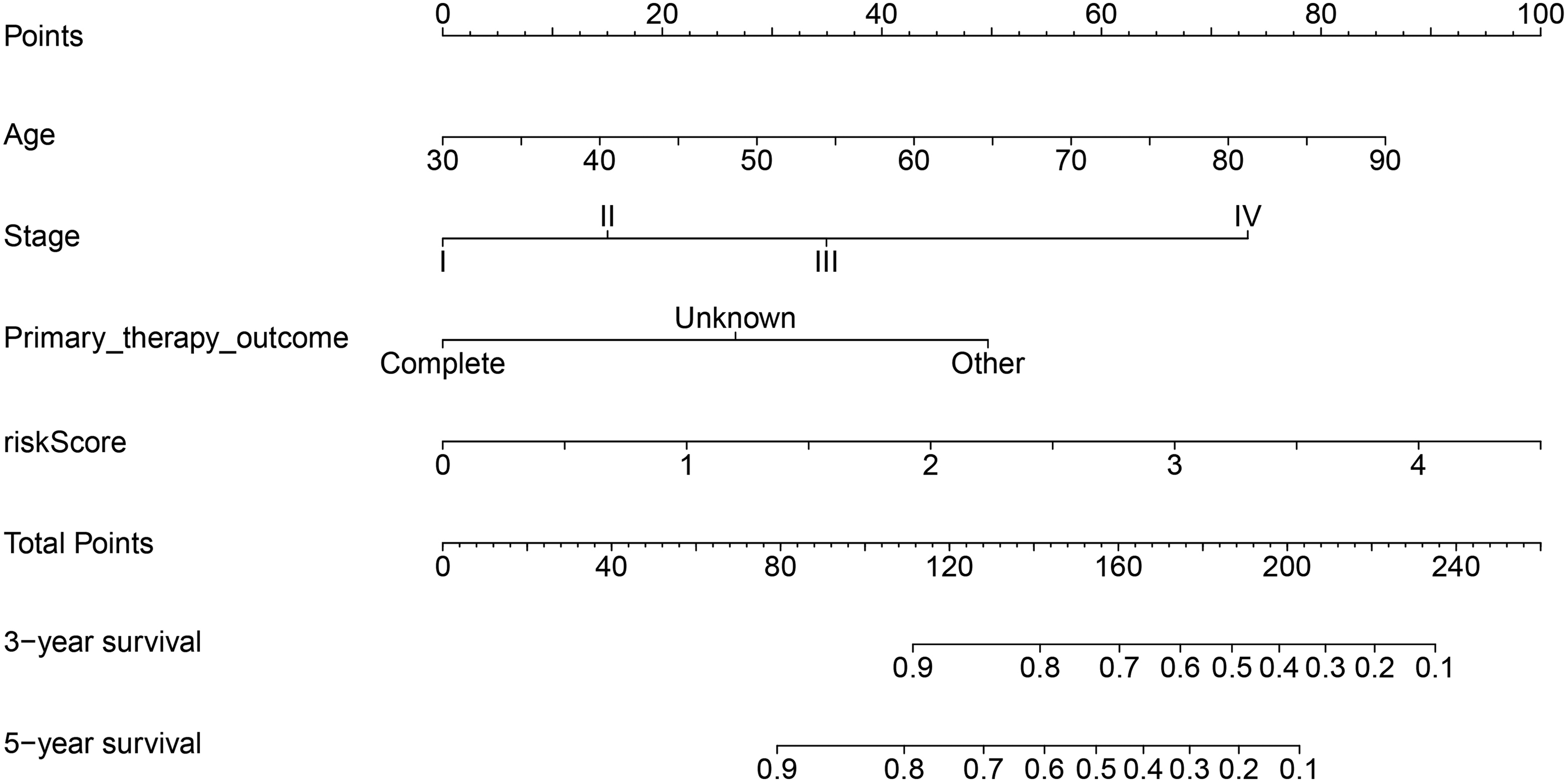
Nomogram for predicting 3- and 5-year overall survival of patients with colorectal cancer.
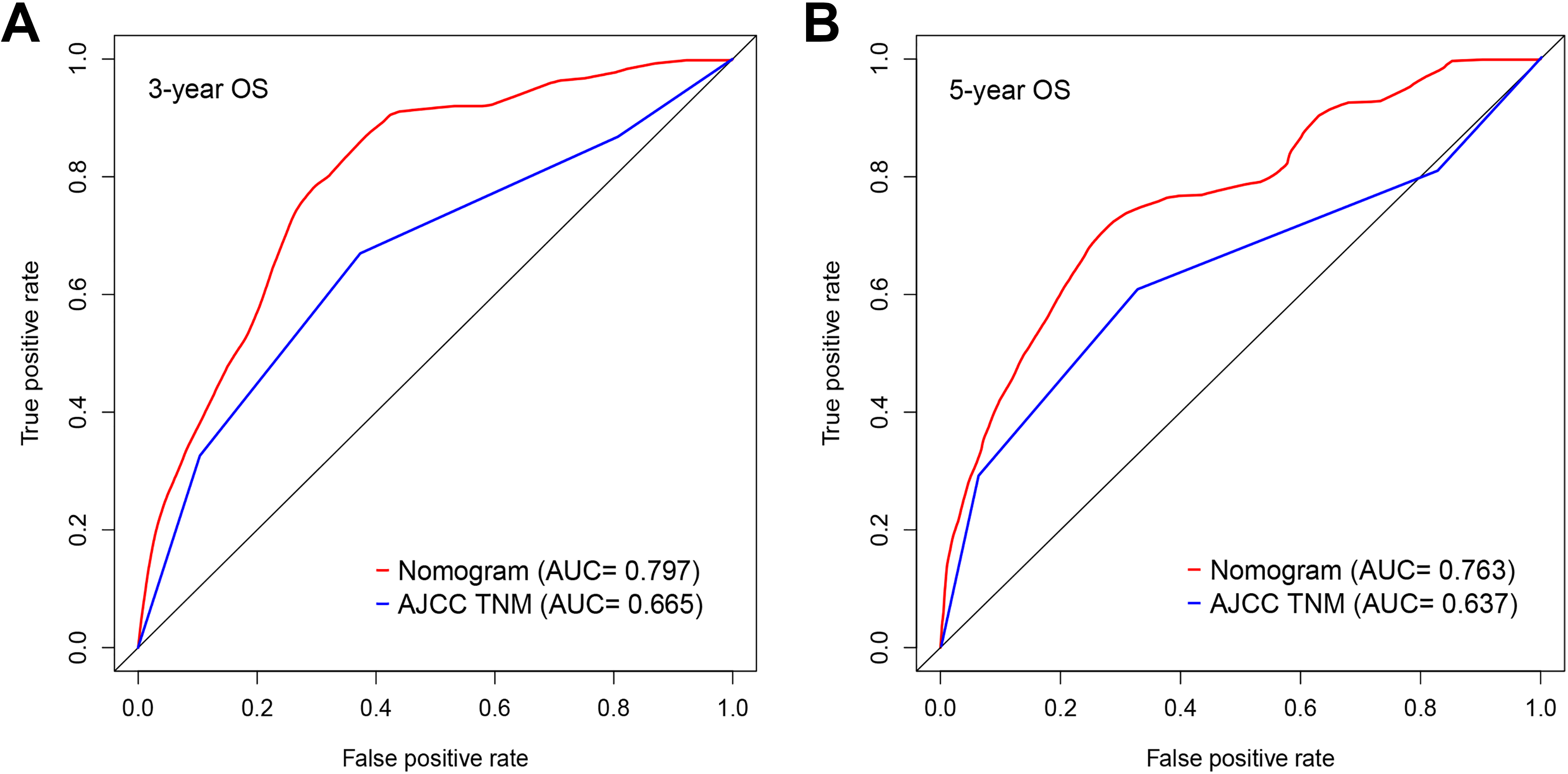
Comparison of the areas under the curve (AUCs) of the nomogram and American Joint Committee on Cancer (AJCC) tumor–node–metastasis (TNM) staging system. A: 3-year OS; B: 5-year OS.

Calibration curves of the nomogram prediction of (A) 3-year and (B) 5-year survival of patients with colorectal cancer.
Discussion
Colorectal cancer remains a huge health burden worldwide with substantial mortality.2-4 The TNM staging system is routinely used as a staging procedure for patients with CRC. However, significant heterogeneity for prognosis and treatment response still exists in the same stage. An accurate prediction of prognosis may inform clinical decisions. In recent years, high-throughput biotechnology is widely used to identify genes marker for CRC. 18 -20 It suggested that a deeper understanding of the genetic characteristics of CRC may lead to a better risk stratification system. Furthermore, many studies have explored the role of autophagy mechanisms in tumorigenesis, including CRC. 21 -23 However, most studies focus on autophagy only by studying a single gene. To our best knowledge, no autophagy-based signature for CRC was constructed. Therefore, we aimed to identify ARGs associated with OS of patients with CRC and establish a novel autophagy-related prognostic model through the TCGA and GEO databases.
In present study, we identified 36 ARGs, which were dysregulated in patients with CRC. Considering these genes may be involved in the initiation of CRC, we further analyzed these different expression ARG functions through the GO and KEGG pathway analysis. The result demonstrated several significantly enriched oncological signatures, such as p53 signaling pathway, apoptosis, HCMV, platinum drug resistance, necroptosis, and ErbB signaling pathway, which might help explain the underlying molecular mechanisms of CRC. After univariate and multivariate analysis, 3 ARGs were used to construct risk score signature using their expression levels weighted by corresponding correlation coefficient. The autophagy-related signature could divide patients into high-risk and low-risk groups based on the median risk score. Patients with high-risk score have significantly worse OS than patients in low-risk group. Furthermore, the predictive accuracy of autophagy-related signature was successfully validated in the cohorts of GSE39582 data set, indicating the good reproducibility of this signature. Multivariate analyses demonstrated that the risk score was independent prognostic factor for patients with CRC. Finally, a prognostic nomogram was established based on risk score and 3 clinicopathologic factors. The prognostic accuracy of the nomogram was confirmed by the discrimination and calibration plots.
Our study has several merits. First, to our knowledge, we firstly established an autophagy-based signature based on multi–data sets. The signature showed favorable predictive ability. Second, the results of the TCGA training data set (mainly composed of the US and European populations) are well validated in the validation set of the Asian population composition. Validation in multiple countries enhances the reliability of the results, indicating that ATG scores may be appropriate for patients of different ethnicities. Third, our nomogram combining risk score with conventional clinical parameters shown significantly improved performance. For example, the nomogram showed a higher predictive ability than the AJCC TNM staging system, consistent with previous studies. 24,25 Moreover, nomogram is of great significance to clinicians and patients because that it can predict individual patient outcomes. When a patient who was diagnosed with a tumor for a few several years previously comes to the clinician for help, he is more concerned about his individual survival risk since at this particular moment rather than the traditional risk since diagnosis.
The proposed autophagy-related signature included 3 ARGs (VEGFA, NRG1, and CDKN2A). All genes in the signature were associated with OS of patients with CRC.
Of these, VEGFA, NRG1, and CDKN2A have been previously reported to have a clear correlation with CRC. Vascular epithelial growth factor A (VEGFA), as a member of the VEGF family, is involved in tumor growth, metastasis, and angiogenesis. 26,27 Several studies indicated that VEGFA is a direct target gene for many microRNAs and was closely related to the initiation and progression of CRC. 28,29 For example, Chen et al 30 demonstrated that miR-150-5p inhibits the progression of CRC by targeting VEGFA in CRC as a tumor suppressor. In addition, the level of VEGF is associated with clinical outcomes. 31 Therefore, VEGF has been the main focus of anticancer drug development. Studies have shown that antiangiogenic therapy can effectively improve the survival rate of patients with CRC, which indicates that inhibition of tumor angiogenesis is a potential method for the treatment of CRC. 32,33 NRG1 is a membrane glycoprotein that mediates cellular signaling and plays a key role in the growth and development of many human organs. 34,35 The gene produces a number of different isoforms by replacing the use of promoters and splicing. 36 These isoforms are expressed in a tissue-specific manner with significant structural differences and are classified into at least 3 subgroups (types I-III). 37 This gene can be used as an oncogene or as a tumor suppressor gene to participate in the development of CRC. 38,39 De Boeck et al 38 found that the expression of NRG1 was significantly upregulated in the CRC tissues and was linked to advanced stage and invasion depth and worse 5-year progression-free survival. NRG1 promotes CRC tumorigenesis by activating the HER2/HER3-dependent PI3K/AKT signaling cascade in CRC cells. On the contrary, there are reports that NRG1 may be the major tumor suppressor gene that causes 8p loss in the colon (short arm of chromosome 8). 39 In our study, we found that NRG1 was downregulated in CRC and associated with worse OS. Therefore, the exact relationship between the NRG1 and CRC needs to be further investigated in future experiments. CDKN2A is one of the original CIMP marker group genes and has important functions in carcinogenesis. 40 Several studies indicated that CDKN2A hypermethylation might be a predictive factor for patients with CRC. 41,42
In our study, functional enrichment analysis indicated that ARGs were identified in several CRC-related pathways including the following: p53 signaling pathway, apoptosis, HCMV, necroptosis, and ErbB signaling pathway. 43,44 P53 is a classic tumor suppressor that often mutates between multiple cancer types. As a key guard in tumorigenesis, p53 is activated in response to carcinogenesis and regulates a large number of genes involved in cell cycle arrest, DNA repair, and apoptosis. 44 Mutations of p53 are highly frequent in CRC, and p53 mutation status is associated with disease outcome. 43 A meta-analysis demonstrated that HCMV infection was associated with an increased risk of CRC. 45 Teo et al 46 found that HCMV infection can enhance cell proliferation, migration, and upregulation of epithelial–mesenchymal transition markers in CRC cells. Taken together, these differentially expressed ARGs are involved in many important CRC-associated biological functions and pathways.
There are still some limitations of this study. First, the sample size is relatively small, and some variables that may be important do not show independent prognostic value. Therefore, future studies with large scale may build more accurate models. Second, due to the lack of primary therapy outcome variable for GSE39582 data set, we are unable to evaluate the predictive accuracy of nomogram by external validation. Third, all analyses in our study are descriptive, and further functional experiments are needed to explore the molecular mechanisms of these 3 ARGs.
Conclusions
We conducted a novel 3 autophagy-related signature to predict OS of patients with CRC, which may help in clinical decision-making for individualized therapy.
Footnotes
Authors’ Note
Z.H. and J.L. contributed equally to this work. Z.H. and S.S.P. designed the experiment. Z.H., J.L., L.L., and P.S. undertook the data acquisition. Z.H., J.L., P.S., J.L., and B.W. were involved in the interpretation of data. Z.H., J.L., L.L., J.Z., and S.S.P. analyzed and visualized the data. All authors drafted and revised the manuscript. The final manuscript was read and approved by all authors. The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Health commission of Hubei Province Scientific Research Project (WJ2019H160).