
Abstract
Acute myeloid leukemia (AML) is a heterogeneous malignancy with diverse genetic mutations and oncogenic pathways influencing treatment response. Despite therapeutic advances, relapse and resistance remain persistent issues. This study integrates genomic and transcriptomic profiling to identify biomarkers of high-risk AML, informing personalized medicine strategies. Nonnegative matrix factorization was applied to RNA sequencing data from the BeatAML cohort (N = 462) for patient subtyping, with survival analysis using the Kaplan–Meier method. Immune profiling via xCell and Gene Set Variation Analysis assessed the tumor microenvironment, with findings validated in the Cancer Genome Atlas AML cohort (N = 173). Using a random forest machine learning model, we developed a 20-gene signature identifying a high-risk subgroup comprising approximately 20% of patients with AML. The high-risk AML subtype was enriched for recurrent FLT3, NPM1, and DNMT3A mutations, activation of PI3K/AKT/mTOR, and complement pathways. Immune profiling revealed an immunosuppressive microenvironment with increased M2 macrophages and mesenchymal stem cells. The 20-gene signature predicted high-risk AML with high accuracy (area under the curve = 0.995, F1 = 0.89). AML cell lines representing high- and low-risk phenotypes identified using the 20-gene signature were tested for drug sensitivity, including the standard-of-care cytarabine, and two targeted therapies, the PI3K inhibitor LY294002 and the MAPK inhibitor selumetinib, selected based on enriched pathways in high-risk AML. High-risk AML cell lines exhibited reduced cytarabine sensitivity but greater responsiveness to PI3K and MAPK/ERK inhibitors, consistent with pathway enrichment results. These findings support molecular stratification and predictive signatures as tools to guide therapy in high-risk AML. Further clinical validation is warranted.
Introduction
Acute myeloid leukemia (AML) is a highly heterogeneous hematological malignancy characterized by the clonal proliferation of immature myeloid cells in the bone marrow, peripheral blood, and other tissues (Graubert and Mardis, 2011; Saultz and Garzon, 2016; Siegel et al., 2024). AML predominantly affects individuals aged 60–70, with a median age at diagnosis of 68 years (Short et al., 2018), and 20,800 estimated new cases in the United States (Siegel et al., 2024).
AML displays considerable genetic diversity and frequently presents with a poor prognosis due to high-risk genetic factors or preexisting conditions such as DNA repair deficiencies, myeloproliferative or myelodysplastic syndromes, and neutropenia (Gabellier et al., 2020; Sekeres and Taylor, 2022). Standard-of-care (SOC) treatment typically includes the nucleoside analogue cytarabine and a topoisomerase II inhibitor such as daunorubicin, both of which are associated with substantial side effects (Hino et al., 2022). These intensive treatments present significant challenges, particularly for older patients who frequently experience rapid disease progression, poor overall health, and a reduced ability to tolerate systemic anticancer therapies (Graubert and Mardis, 2011).
Immunotherapy has also emerged as a promising modality in AML treatment, leveraging the immune system’s ability to target and eliminate leukemic cells (Grosso et al., 2015). Treatments such as allogeneic hematopoietic stem cell transplantation (allo-HSCT) remain the most successful immune-based therapies for AML, particularly with advancements in alternative donors’ use (Grosso et al., 2015; Kantarjian et al., 2021). While allo-HSCT therapies have prolonged long-term survival in many cases, the treatment-related toxicity such as graft-versus-host disease (McDonald et al., 2020) and relapse after treatment remain as major challenges.
Many new treatments have been developed in the last decade for AML. Targeted therapies, such as midostaurin, a FLT3 inhibitor (Zhao et al., 2022), and enasidenib, an IDH2 inhibitor (Stein et al., 2017), have demonstrated efficacy in specific genetic subtypes of AML. Additionally, the reapproval of gemtuzumab ozogamicin, an antibody-drug conjugate targeting CD33 (Kantarjian et al., 2021), and the novel liposomal formulation CPX-351 for therapy-related AML (Lancet et al., 2018), reflect the growing role of targeted approaches in treating this disease. The PI3K/AKT/mTOR and RAS/MEK/ERK signaling pathways are critical to AML pathogenesis, mediating key processes such as cell proliferation, survival, and differentiation. Targeting these pathways represents a promising therapeutic strategy, with ongoing preclinical studies and clinical trials actively exploring their potential in AML treatment (Carter et al., 2020; Li et al., 2022; Tseng et al., 2023).
Despite recent advancements in the treatment landscape, AML remains a significant challenge, particularly among elderly patients and those with relapsed or refractory AML. Moreover, many elderly patients cannot undergo intensive therapy due to age-related factors, poor overall health, or other medical conditions, and the disease often relapses or becomes refractory, occurring in 30–40% of cases (Graubert and Mardis, 2011; Lohse et al., 2018; Saultz and Garzon, 2016; Stratmann et al., 2022). This leads to overall poor survival outcomes, with only 31% of patients with AML surviving 5 years postdiagnosis (Lohse et al., 2018). All in all, the clinical heterogeneity of AML necessitates precise classification systems to effectively guide treatment decisions and predict patient prognosis (Wang et al., 2022).
The development of high-throughput sequencing technologies has profoundly transformed our understanding of the molecular landscape of AML (Grosso et al., 2015; Wang et al., 2022). Comprehensive genomic studies have unveiled a myriad of recurrent mutations, chromosomal aberrations, and epigenetic modifications that drive AML pathogenesis (Kantarjian et al., 2021). Notable mutations in genes such as FLT3, NPM1, and DNMT3A have been linked to specific clinical outcomes and therapeutic responses (Graubert and Mardis, 2011; Mevatee et al., 2017). The tumor microenvironment (TME), particularly the immune landscape, plays a critical role in AML progression and treatment response (Mevatee et al., 2017). Immune cells within the bone marrow and peripheral blood engage in dynamic interactions with leukemic cells, influencing their growth, survival, and resistance to therapies (Graubert and Mardis, 2011; Mevatee et al., 2017). Understanding these immune interactions is essential for developing novel immunotherapeutic approaches and improving patient outcomes. However, integrating these diverse molecular data into cohesive, clinically actionable frameworks remains a formidable challenge. Immune profiling, combined with genomic and transcriptomic analyses, offers a comprehensive approach to elucidating the complex biology of AML and identifying potential therapeutic targets (Stratmann et al., 2022).
To address the challenge due to clinical and molecular heterogeneity of AML, we investigated whether integrated genomic and immune transcriptomic profiling could reveal distinct high-risk subgroups with underlying genes enriched in biological pathways. Building on this, we aimed to define a robust gene signature associated with poor prognosis, with the potential to inform risk stratification and guide therapeutic strategies.
Materials and Methods
Data sources
This study employed clinical features, mutation, and RNA sequencing (RNA-seq) datasets from two primary patient cohorts: BeatAML (N = 462) and the cancer genome atlas acute myeloid leukemia (TCGA-LAML) cohort (N = 173), as well as gene expression data from AML cell lines in the Cancer Cell Line Encyclopedia (CCLE).
The BeatAML cohort is a large, publicly available dataset derived from the BeatAML master trial, which profiled AML patient samples using comprehensive genomic characterization and ex vivo drug sensitivity screening to support precision medicine approaches (Tyner et al., 2018). Controlled-access genomic data were obtained via the National Center for Biotechnology Information’s Database of Genotypes and Phenotypes (dbGaP, Study ID: phs001657.v2.p1), and an overview of the dataset is available at https://www.cancer.gov/ccg/blog/2019/beataml.
The TCGA-LAML dataset was accessed from TCGA via the Genomic Data Commons portal (https://portal.gdc.cancer.gov/). Gene expression data for AML cell lines were obtained from the CCLE through the Broad Institute’s portal (https://sites.broadinstitute.org/ccle/).
The study employed in vitro cell lines and publicly available clinical and omics data and did not require informed consent or research ethics board approval.
Patient stratification by clustering analysis
To stratify patients into distinct molecular subtypes, we utilized nonnegative matrix factorization (NMF; Frigyesi and Hoglund, 2008), an unsupervised machine learning algorithm, on the BEATAML RNA-seq data (N = 462; Tyner et al., 2018) to identify transcriptomic-based molecular subtypes. We focused on the top 1000 most variable genes across the datasets. The high-risk group identified in the BeatAML cohort was subjected to the same clustering and analysis methods in an independent validation dataset, which is the TCGA-LAML cohort (Ley et al., 2013).
Clinical outcomes
Survival analysis evaluated differences in overall survival (OS) between the high-risk and the low-risk patients using the Kaplan–Meier method with statistical differences assessed by the log-rank test (Jager et al., 2008). Survival curves and hazard ratios (HRs) were generated using the survival and survminer packages in R program. A p value <0.05 was considered statistically significant.
Genomic data analysis for AML subtypes
To evaluate mutational differences across AML subgroups, mutation data were visualized by using OncoPrints, which provide a compact graphical summary of mutation types (e.g., missense, nonsense, and frameshift) across multiple genes and samples. These plots allow for easy comparison of mutation frequency and co-occurrence across clusters. The OncoPrints were generated using mutation annotation format data derived from the BeatAML and TCGA-LAML cohorts. Mutation frequency differences across clusters were statistically evaluated using Fisher’s exact test.
Differential gene expression and pathway analysis
To identify differentially expressed genes between AML samples from Cluster 3 and those from other clusters, we utilized the DESeq2 package in R program (Version 4.4.3). We filtered genes for analysis by retaining those with a total count of at least 100 across all samples to ensure sufficient expression levels for reliable statistical evaluation. Genes were considered differentially expressed based on a threshold of a Log2 |Fold Change| >2 and a stringent adjusted p value (false discovery rate) of <0.05.
Pathway analysis was conducted through Gene Set Variation Analysis (GSVA) using transcriptome data to explore the functional landscape across different molecular subtypes identified in patients with AML. We utilized established gene set collections from three major pathway databases: Hallmark (Liberzon et al., 2015), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis (Kanehisa and Goto, 2000), and Reactome (Matthews et al., 2009) to identify pathways enriched in each subtype. Raw RNA-seq counts were normalized using the variance-stabilizing transformation method from the DESeq2 package. GSVA scores were computed for each sample using the normalized expression data. Differences in pathway activities were assessed using a linear modeling approach via the limma (Ritchie et al., 2015) R package.
Immune characterization
The immune characterization analysis was conducted using the xCell method (Aran et al., 2017), combined with KEGG and REACTOME immune signatures evaluated through GSVA scores. Significantly different immune signatures, identified through stringent statistical thresholds, were further analyzed to pinpoint specific cell types or pathways that might be driving the immune characteristics of high-risk AML subtypes. Statistical comparisons of immune signature enrichment scores across clusters were performed using pair-wise Wilcoxon rank-sum tests, and comparisons with p < 0.05 were considered statistically significant.
Predictive model development
The BeatAML cohort was applied to develop and test a predictive model and to generate a gene signature indicative of high-risk subgroup patients. The validation cohort, consisting of gene expression data from the TCGA database, was used to assess the generalizability and effectiveness of the derived gene signature across different populations.
The BeatAML dataset was split into training and test sets in an 80:20 ratio to develop a predictive model for classifying patients into the high-risk subgroup. We evaluated the following machine learning algorithms: naive Bayes, decision tree, random forest, support vector machine (SVM), and K-nearest neighbor (KNN) using the top 20 genes identified as key features for the high-risk subgroup (Ahmed et al., 2017; Carracedo-Reboredo et al., 2021; Diaz-Uriarte and Alvarez de Andres, 2006). These models were implemented in R program (version 4.4.3) using the caret and randomForest packages. Each model’s performance was assessed through receiver operating characteristic (ROC) curve analysis, confusion matrix, accuracy, recall, precision, and F1-score. The best model was selected based on the performance metrics, and the 20 most significant genes were used to develop a predictive gene signature for the high-risk subgroup.
Validation of 20-gene signature in patient cohort
The predictive performance of the 20-gene signature was first assessed within the test portion of the BeatAML cohort and subsequently validated using an independent TCGA dataset. This approach ensured that the model was effective not only within the cohort it was developed on but also robust across different datasets. Evaluation metrics such as ROC curves, accuracy, precision, recall, and F1-scores were calculated to assess the model’s effectiveness. Additionally, GSVA was used to calculate 20-gene signature enrichment scores for each patient in this validation cohort. These scores were then utilized to identify patients with high signature enrichment, particularly focusing on the identification of patients within high-risk subgroup.
Identification of high-risk like cell lines and drug sensitivity analysis by using 20-gene signature
To define high-risk like cell lines, the 20-gene signature was applied using the random forest classifier. The model’s predictions were validated by comparing them with GSVA (Hanzelmann et al., 2013) scores, and high and low groups were established based on these classifications.
To assess the drug sensitivity of AML cell lines characterized by the high-risk 20-gene signature, IC50 values and area under the curve (AUC) metrics for various inhibitors were obtained from the Cancer Therapeutics Response Portal (CTRP) (Rees et al., 2016). This portal provides comprehensive drug response data, which were utilized to correlate the effectiveness of different inhibitors with the gene signature scores of the cell lines. Spearman rank correlation analysis was used to evaluate associations between signature scores and AUC values. The analysis involved calculating correlation coefficients to determine which drugs showed enhanced sensitivity in cell lines that exhibited high scores on the 20-gene signature. This approach aims to identify inhibitors that are particularly effective against AML cell lines that resemble the high-risk, Cluster3 subgroup, potentially guiding targeted therapy development for this challenging patient cohort. All statistical analyses were conducted using R program (R.4.4.3), with data processing carried out through the R Bioconductor package (https://www.bioconductor.org). A p value <0.05 was considered significant. Additionally, Colab notebook and Python 3.12.4 were utilized for IC50-AUC analysis.
In vitro validation experiments
Cell culture
AML cell lines were obtained from the following resources: THP-1 (ATCC, Manassas, VA, USA; TIB-202), KG-1 (ATCC, Manassas, VA, USA; CCL-246), MV-4-11 (ATCC, Manassas, VA, USA; CRL-9591), and AML-193 (DSMZ, Braunschweig, Germany; ACC 549).
THP-1 was cultured in RPMI 1640 (Gibco, Thermo Fisher Scientific, Waltham, MA, USA; A10491) with 10% donor bovine serum with iron (Gibco, 20371-030) and 0.1% 2-mercaptoethanol (Gibco, 31350-010). KG-1 was cultured in IMDM (Capricorn, Ebsdorfergrund, Germany, IMDM-A) and 20% donor bovine serum with iron (Gibco, 20371-030). MV-4-11 was cultured in IMDM (Capricorn, IMDM-A) and 10% fetal bovine serum (ATCC, 30-2020) and AML-193 was cultured in IMDM (Capricorn, IMDM-A) with 10% donor bovine serum with iron (Gibco, 20371-030) and 0.5% human GM-CSF recombinant protein (Immunotools, Friesoythe, Germany, 11343125). All cell lines were cultured at 37°C, 5% CO2, and ambient oxygen level (20%).
Reagents
Cytarabine (Selleckchem, Houston, TX, USA, S1648) was prepared as a stock solution in Phosphate-buffered saline (PBS) at a concentration of 10 mM and stored at −20°C. Working dilutions were prepared freshly in cell line specific medium to achieve final concentrations as needed for each experiment.
PI3K inhibitor LY294002 (MedChemExpress, Monmouth Junction, NJ, USA, HY-10108) was prepared as a 100 mM stock solution in dimethyl sulfoxide (DMSO) and stored at −20°C. For each experiment, the stock solution was diluted in cell culture medium to achieve the desired working concentration.
MAPK inhibitor selumetinib (MedChemExpress, HY50706A) was prepared as a 100 mM stock solution in DMSO and stored at −20°C. Immediately before use, the stock solution was diluted in cell culture medium to the required final concentration. The final DMSO concentration was maintained constant across all conditions, and control wells contained an equivalent amount of DMSO to match the highest concentration used in the treatment groups.
Cell viability reagent CellTiter-Glo (Promega, Madison, WI, USA) was used to measure cell viability according to manufacturer’s instructions.
Cell viability assay
Adherent target cells were detached using trypsin, collected, and cell concentration and viability were determined. Cells were plated at 40,000 cells per well (20.000 for THP-1) and were cultured for 24 h at 37°C in a 5% CO2 incubator. Fifty microliters containing either cytarabine, the PI3K inhibitor LY294002, or the MAPK inhibitor selumetinib in the described concentrations was added to wells containing 100 µL of target cell suspension. Plates were incubated for 72 h at 37°C, 5% CO2. After incubation, cell viability was assessed using the CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI, USA). An equal volume of CellTiter-Glo reagent was added to each well, and the plate was shaken for 2 min to induce cell lysis and incubated at room temperature for 10 min. Luminescence was measured using a microplate reader. Viability was calculated by normalizing luminescence values to those of untreated control wells.
Results
Patient stratification
To identify clinically relevant subgroups within AML, NMF was applied to transcriptomic data from the BeatAML dataset, revealing five distinct clusters with significant differences in OS. Kaplan–Meier survival analysis (Fig. 1a) demonstrated that Cluster 3, characterized by the worst prognosis, had a median survival of 348 days, whereas Cluster 4, associated with the best prognosis, had a median survival of 1237 days, revealing the substantial differences in clinical outcomes between these clusters (log-rank p = 0.017). A Cox proportional hazard model comparing Cluster 3 to all other clusters (Fig. 1b) showed a HR of 1.41 (95% confidence interval [CI]: 1.03–1.92, p = 0.03), indicating that Cluster 3 contained a significantly higher risk subpopulation of patients with AML.
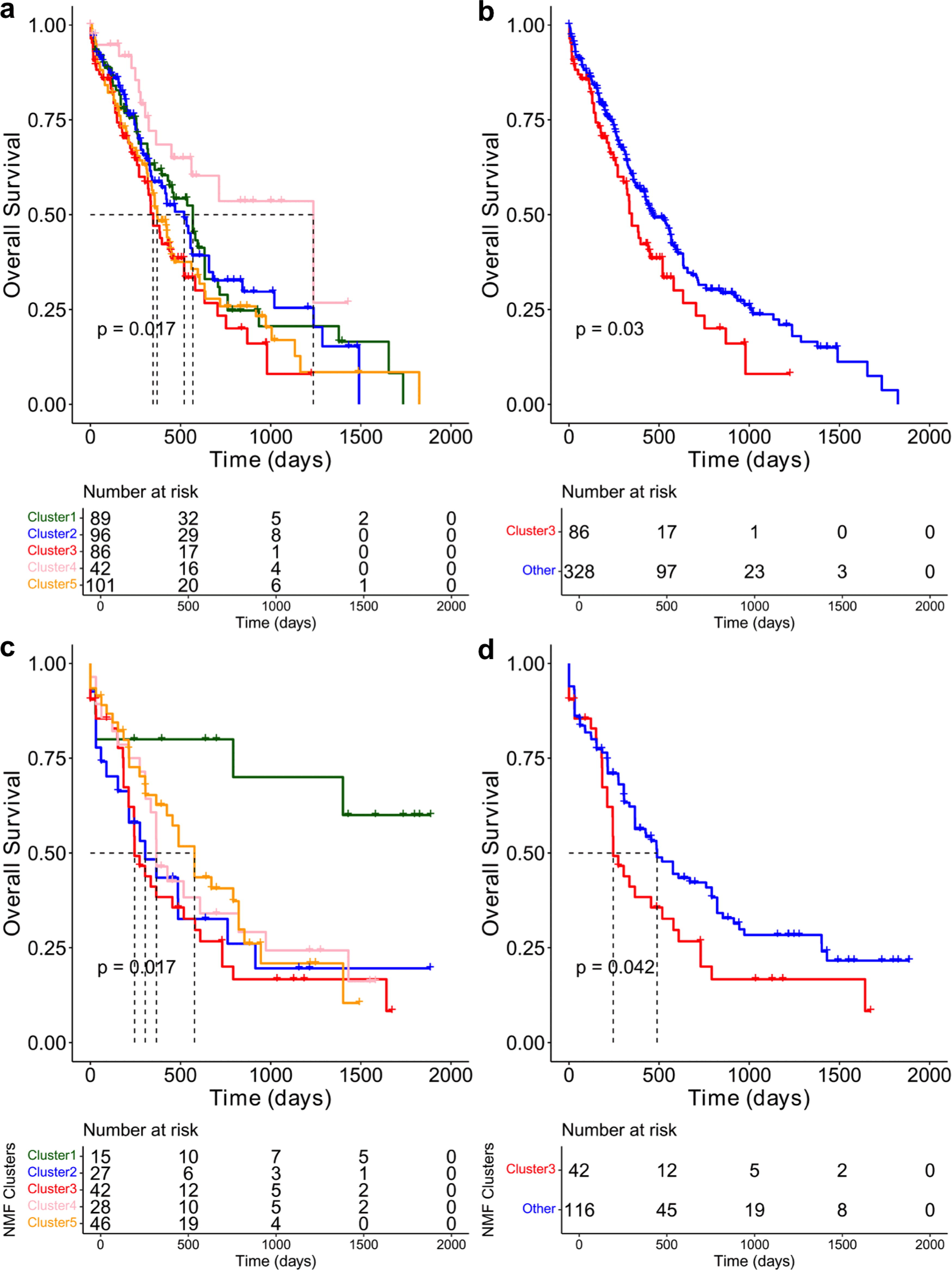
Overall survival of AML patient subpopulations. Kaplan–Meier survival curves comparing transcriptomic clusters.
Survival analysis for the TCGA dataset validated the findings from the BeatAML dataset (Fig. 1c). In this cohort, Cluster 3, characterized by the worst prognosis, had a median survival of 245 days, whereas median had not been reached in Cluster 1, and the Cluster 5 showed much longer survival (median = 577 days), highlighting significant survival differences between the clusters (log-rank p = 0.02). Comparing Cluster 3 with the other groups (Fig. 1d) again demonstrated Cluster 3 as a significantly higher risk patient subgroup (HR = 1.54, 95% CI: 1.01–2.33, p = 0.04). These results corroborated the poor prognostic outcome for Cluster 3, further supporting its association with unfavorable clinical outcomes across datasets.
Genomic features of high-risk patients
To identify the unique genomic characteristics driving poor outcomes in the high-risk subgroup, mutation profiles across all clusters in the BeatAML cohort were analyzed. Cluster 3, previously identified as the high-risk subgroup based on survival analysis, was characterized by significantly shorter OS. Mutations in FLT3 (25%), NPM1 (21%), and DNMT3A (19%) were among the most frequent across all clusters, whereas mutations in TET2 (12%), NRAS (13%), and RUNX1 (10%) were also present, reflecting the overall genetic diversity observed (Fig. 2a). The high-risk subgroup, specifically, had FLT3 (34.4%), NPM1 (29.5%), and DNMT3A (16.4%) mutations, with 6.56% of these patients exhibiting all three mutations concurrently; details are provided in Supplementary Table S1. Fisher’s exact test revealed a significantly higher rate of combined mutations in all three genes (FLT3, NPM1, and DNMT3A) in Cluster 3 compared with Clusters 2 (p = 0.03) and 5 (p = 0.035). Broader analysis of patients with at least two of the three mutations showed that Cluster 3 had fewer such cases than Cluster 1 (p = 0.0005) but more than Clusters 2, 4, and 5, signifying Cluster 3′s distinct mutation profile (Fig. 2a).
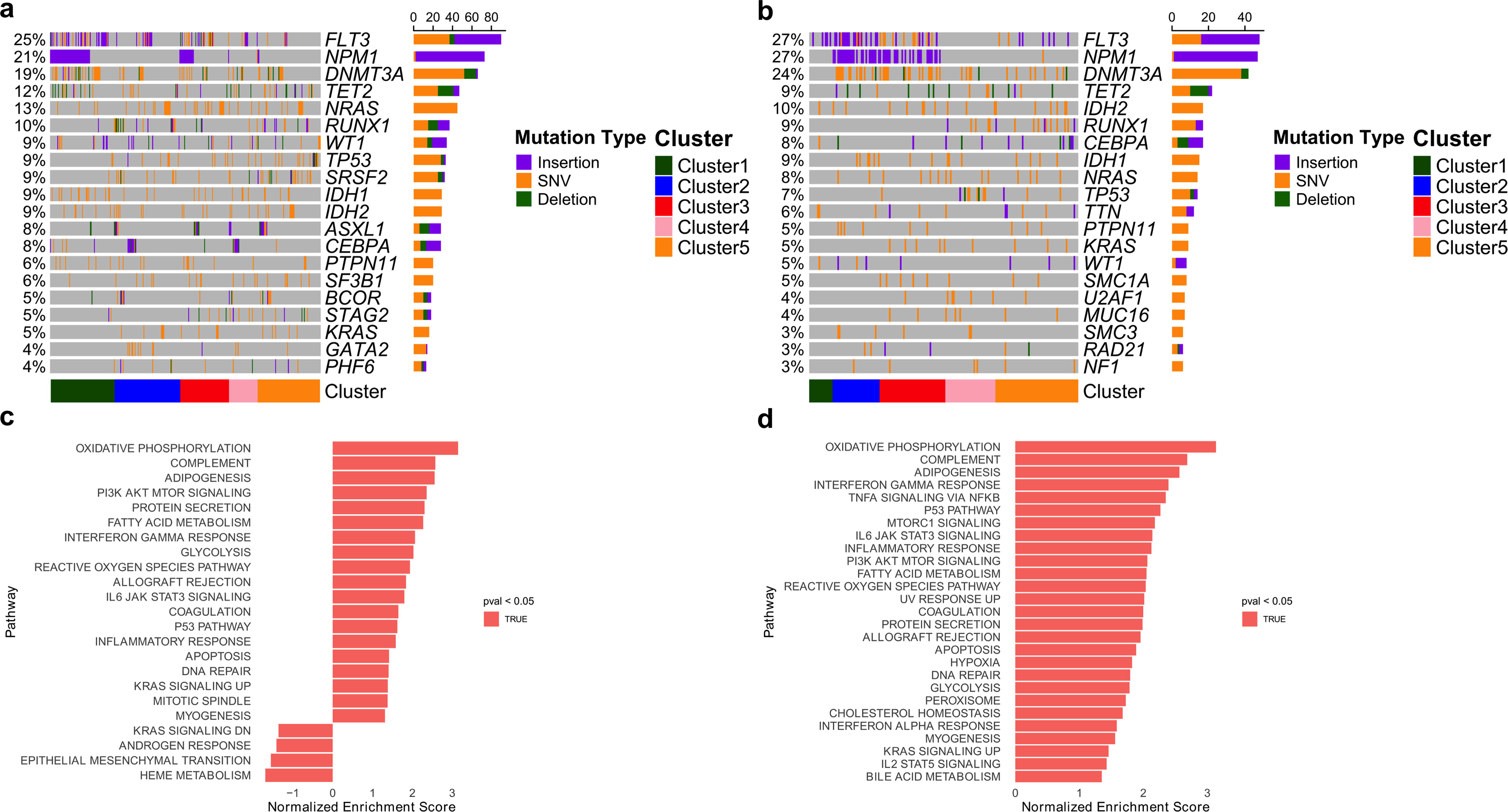
Mutation landscape of patients with AML and genomic features of the poor prognostic set.
In the TCGA validation cohort, FLT3 and NPM1 mutations were observed in 27% of patients, DNMT3A in 24%, with additional TET2, IDH2, and RUNX1 mutations at lower frequencies in high-risk patients, similar to BeatAML (Fig. 2b). The high-risk subgroup, specifically, had FLT3 (35.7%), NPM1 (54.8%), and DNMT3A (38.1%) mutations. Notably, 16.7% had all three mutations, a significantly higher rate compared with other subpopulations, including Clusters 4 (p = 0.017) and 5 (p = 0.0024) with details provided in Supplementary Table S2. Fisher’s exact test also indicated high-risk subgroup, Cluster 3, had significantly more patients with at least two mutations compared with Clusters 1, 4, and 5, reinforcing Cluster 3′s unique mutation burden among high-risk AML patients.
Cluster 3 high-risk patients were found to harbor key mutations in FLT3, NPM1, and DNMT3A genes. These mutations are frequently associated with poor prognosis and were accompanied by the activation of several oncogenic pathways, including PI3K/AKT/MTOR, TGFB, and NOTCH signaling (DiNardo and Cortes, 2016). The presence of these genetic alterations emphasizes the aggressive nature of AML in Cluster 3 and provides potential targets for therapeutic intervention.
Transcriptomic features of poor prognostic set of patients
To characterize the transcriptomic features of high-risk patients in Cluster 3, GSVA was performed and hallmark gene sets were curated to identify pathways enriched in this subgroup. In the BeatAML Cluster 3, GSVA using HALLMARK gene sets revealed enrichment of several key pathways, including oncogenic pathways such as KRAS signaling (p = 0.005), PI3K/AKT/mTOR signaling (p = 0.00088), and IL6/JAK/STAT3 signaling (p = 0.00083), as well as immune system-related pathways such as interferon gamma response (p = 0.0012), inflammatory response (p = 0.0013), and complement activation (p = 0.0012). These findings highlighted the role of both oncogenic signaling and immune-related mechanisms in contributing to poor outcomes in BeatAML patients (Fig. 2c).
The TCGA validation cohort confirmed the elevated pathways linked to high-risk AML. Among the most enriched pathways were oxidative phosphorylation (p = 5.9 × 10−5), complement activation (p = 6.0 × 10−5), and interferon gamma response (p = 0.0002), emphasizing the role of metabolic and immune system alterations. Additionally, oncogenic signaling pathways were enriched, further supporting their role in disease progression (Fig. 2d). These findings were consistent with the BeatAML results, emphasizing the impact of immune dysregulation and oncogenic signaling in high-risk AML.
Immune signatures of poor prognostic patient subpopulation
To further characterize the immunological features of high-risk patients, immune pathway and cell-type enrichment scores were visualized using boxplots to compare distributions across clusters. Statistical differences were evaluated using nonparametric tests to assess pair-wise differences, and significance was annotated directly on the plots (Fig. 3a–h). The Cluster 3 high-risk patients demonstrated a notably increased immune suppressive TME compared with the other clusters. Immune profiling using xCell scores and GSVA-based pathway analysis, both of which are derived from transcriptomic data, revealed that Cluster 3 exhibited significantly elevated expression-based scores for complement pathway activation, Fc-gamma receptor (FCGR)-mediated phagocytosis, and specific immune-related pathways, along with an increased presence of M2 macrophages, monocytes, and mesenchymal stem cells (MSCs). Notably, the ImmuneScore, a composite metric from xCell that reflects the overall immune cell infiltration based on enrichment of multiple immune cell types, was also significantly higher in Cluster 3, further supporting the presence of a more immune-infiltrated and immunosuppressive TME. These results were derived from transcriptomic signatures rather than traditional methods such as flow cytometry or immunohistochemistry. This distinct immune activation and cellular composition that are not cytotoxic against tumor highlight high-risk patients’ unique immunological landscape, distinguishing it from other clusters. These elevated immune signatures of high-risk patients were validated within the TCGA cohorts as well (Supplementary Fig. S1).
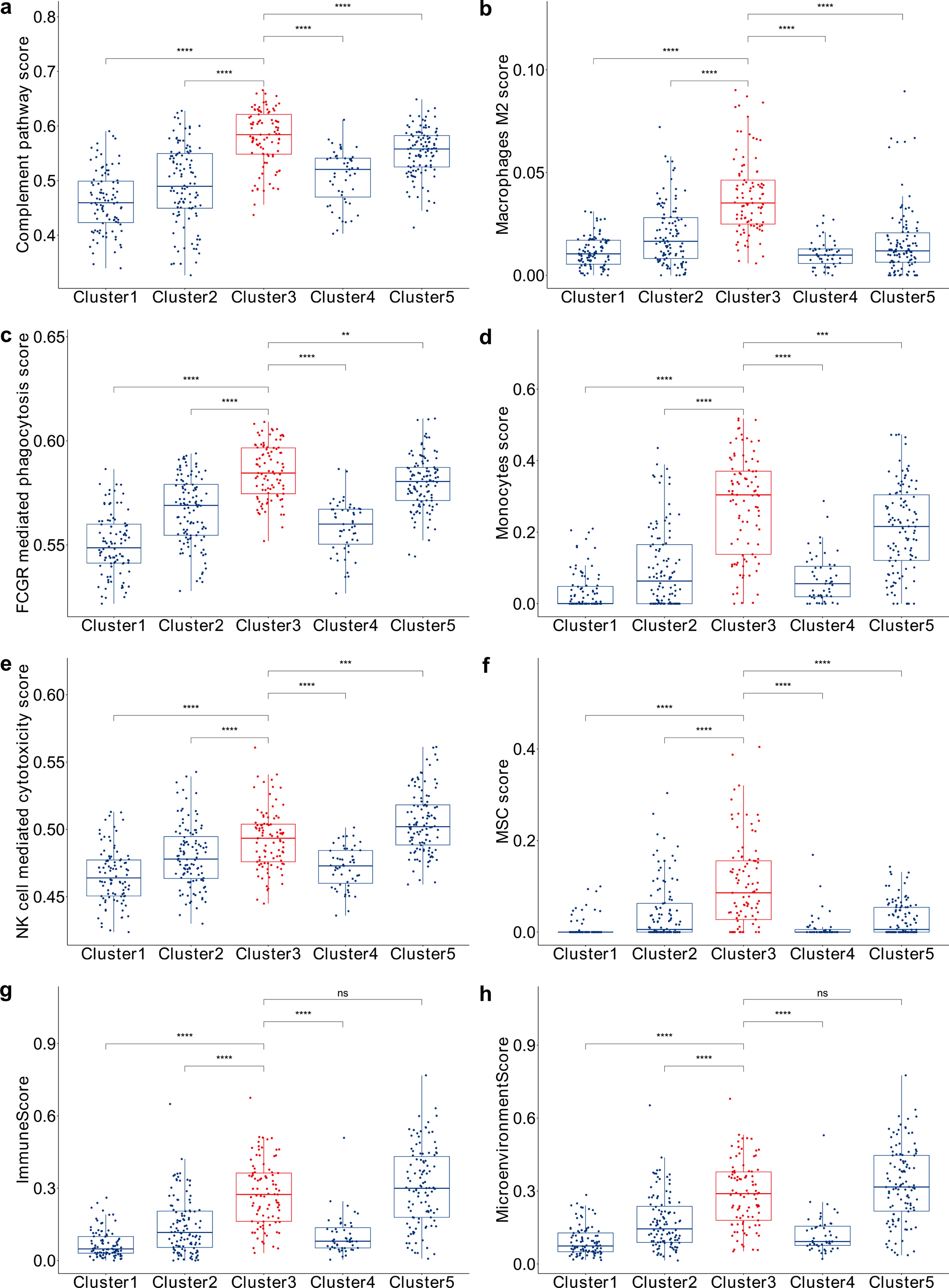
Comparative analysis of immune pathway scores across different clusters in BeatAML. The distribution of pathway scores for
These elevated immune features suggest a complex interplay between AML cells and the immune system, contributing to the poor prognosis of this subgroup (Khaldoyanidi et al., 2021; Mendez et al., 2019). Specifically, the increased complement pathway activation and FCGR-mediated phagocytosis indicate enhanced immune mechanisms aiding leukemic cell survival (Kolev et al., 2011). The higher NK cell activity reflects an ongoing immune response, whereas the enrichment of M2 macrophages suggests an immunosuppressive microenvironment that supports tumor growth (Baginska et al., 2013). Additionally, the elevated presence of monocytes indicates a dynamic and potentially dysregulated immune environment, whereas the higher MSC activity highlights complex stromal-immune interactions facilitating leukemia progression (Antoon et al., 2024). These findings reveal the distinct immune suppressive microenvironment of high-risk AML patients, characterized by enhanced complement activation, FCGR-mediated phagocytosis, increased M2 macrophages, monocytes, and MSCs. This immune landscape appears to contribute to the adverse clinical outcomes associated with this subgroup.
Establishing a 20-gene signature for predicting poor prognosis
High-risk patients with AML, such as those in Cluster 3, have significantly worse clinical outcomes (Fig. 1), highlighting the need for advanced tools to predict poor prognosis and inform targeted therapeutic approaches. Although clustering analyses identified these subgroups, additional steps are required to translate these findings into clinically actionable strategies. The development of a focused gene signature addresses this critical gap by providing a reliable and interpretable approach to consistently stratify high-risk patients and identify those with poor prognoses, emphasizing the importance of robust methodologies to improve risk assessment and guide personalized treatment strategies.
To address this, we developed a 20-gene prognostic signature using machine learning. Five models (naive Bayes, decision tree, random forest, SVM, and KNN) were evaluated for classifying high-risk cases (Fig. 4a). The confusion matrix (Fig. 4b) summarizes the model’s classification performance by comparing predicted and actual class labels, showing the number of true positives, false positives, true negatives, and false negatives. Model performance metrics including accuracy, precision, recall, F1-score, and AUC are summarized in Supplementary Table S3. Random forest outperformed others in training data (accuracy: 95.7%, recall/precision: 89.5%, and AUC: 0.98), with feature importance analysis identifying 20 genes critical for risk stratification (Supplementary Table S3).
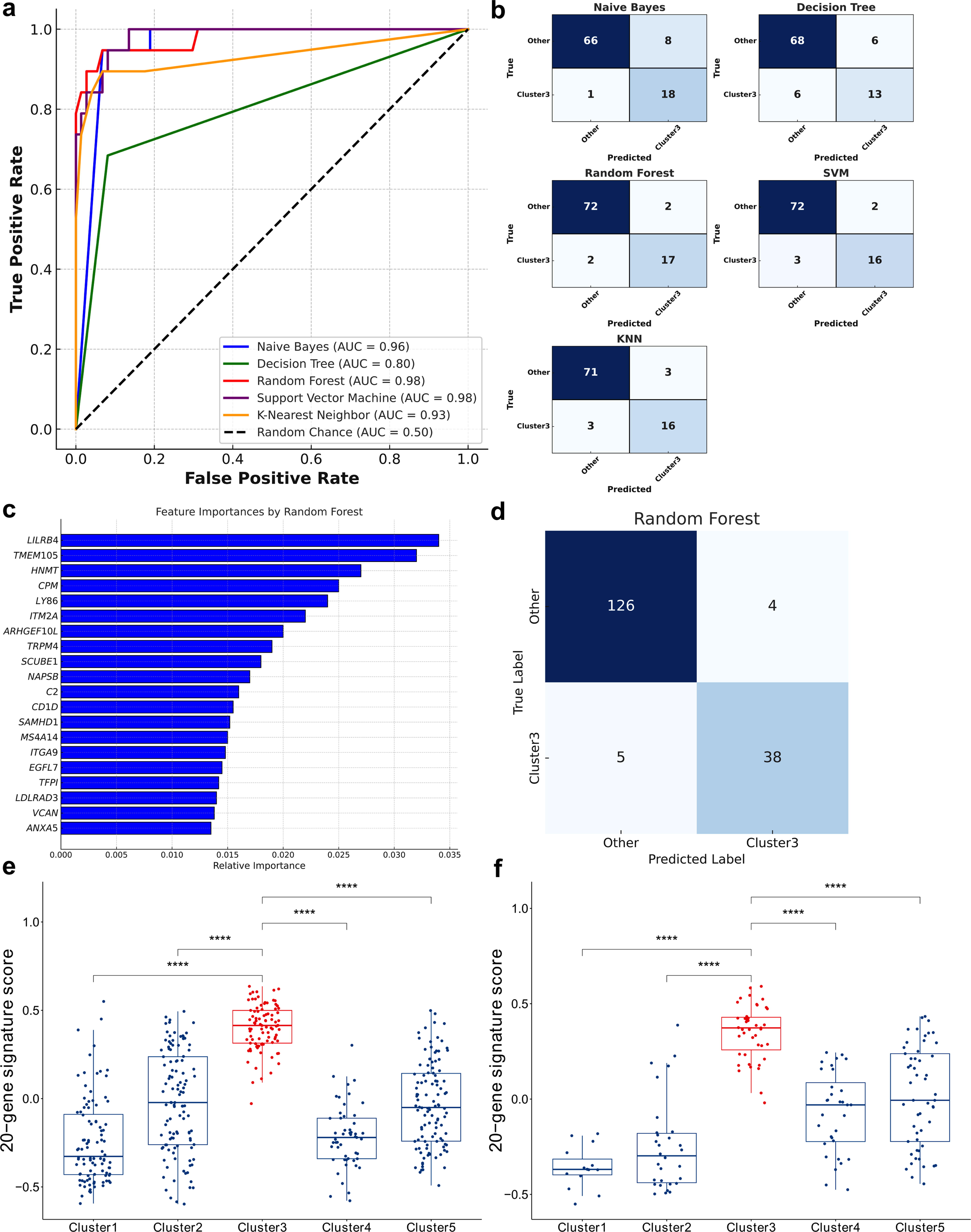
Analysis of predictive model performance for high-risk subgroup classification.
Gene importance in the random forest model was determined based on the mean decrease in Gini impurity, which reflects how much each gene contributed to improving the model’s classification accuracy. The top 20 genes were selected based on the highest importance scores (Fig. 4c). Validation in the TCGA cohort confirmed robust performance (accuracy: 94.8%, AUC: 0.995, and precision/recall: 0.90/0.88; Fig. 4d). GSVA further validated the signature’s ability to distinguish high-risk Cluster 3 in BeatAML and TCGA datasets (Fig. 4e–f).
The 20-gene signature (Fig. 4c) includes critical genes such as LILRB4, CD1D, and TRPM4, which are associated with the regulation of T cell immunity (Bendelac et al., 2007; Deng et al., 2018; Launay et al., 2004). Additionally, genes such as SAMHD1, C2, ITM2A, LY86, and ITGA9 play crucial roles in immune regulation (Rehwinkel et al., 2013; Ricklin et al., 2016; Sheedy et al., 2010), whereas ANXA5 and SCUBE1 (Lin et al., 2023; Peng et al., 2014) are involved in coagulation processes. ITGA9 and TRPM4 contribute to cell proliferation and survival (Armisen et al., 2011; Wu et al., 2022), with VCAN also functioning as a checkpoint protein (Hope et al., 2017), highlighting its potential importance in patient outcomes. This signature also encompasses genes such as TMEM105, HNMT, CPM, ARHGEF10L, NAPSB, MS4A14, EGFL7, LDLRAD3, and TFPI, representing diverse biological pathways that contribute to poor prognosis.
This 20-gene signature serves as a robust and interpretable tool for identifying high-risk AML patients and guiding therapeutic strategies. Its application in this study enables the identification of high-risk cell lines and facilitates in vitro drug screening, providing a foundation for further research and clinical translation.
Profiling high-risk AML cell lines with a 20-gene signature
As a next step, AML cell lines were identified that exhibited characteristics similar to those of high-risk Cluster 3 patients. A 20-gene signature was applied to the 34 AML cell lines (Cancer Cell Line Encyclopedia and Genomics of Drug Sensitivity in Cancer, 2015) using a random forest classifier (Fig. 5a). This approach enabled the effective discrimination of cell lines within the CCLE that mirrored the transcriptomic profile of the high-risk subgroup identified in AML patients, providing a valuable resource for further research and potential therapeutic targeting in AML. Cell lines including THP1, NOMO1, MONOMAC1, OCIAML3, OCIAML2, EOL1, OCIAML5, MV411, SIGM5, and PL21 were identified as representative of the high-risk subgroup based on their ranking within the top 10 AML cell lines by 20-gene signature score.
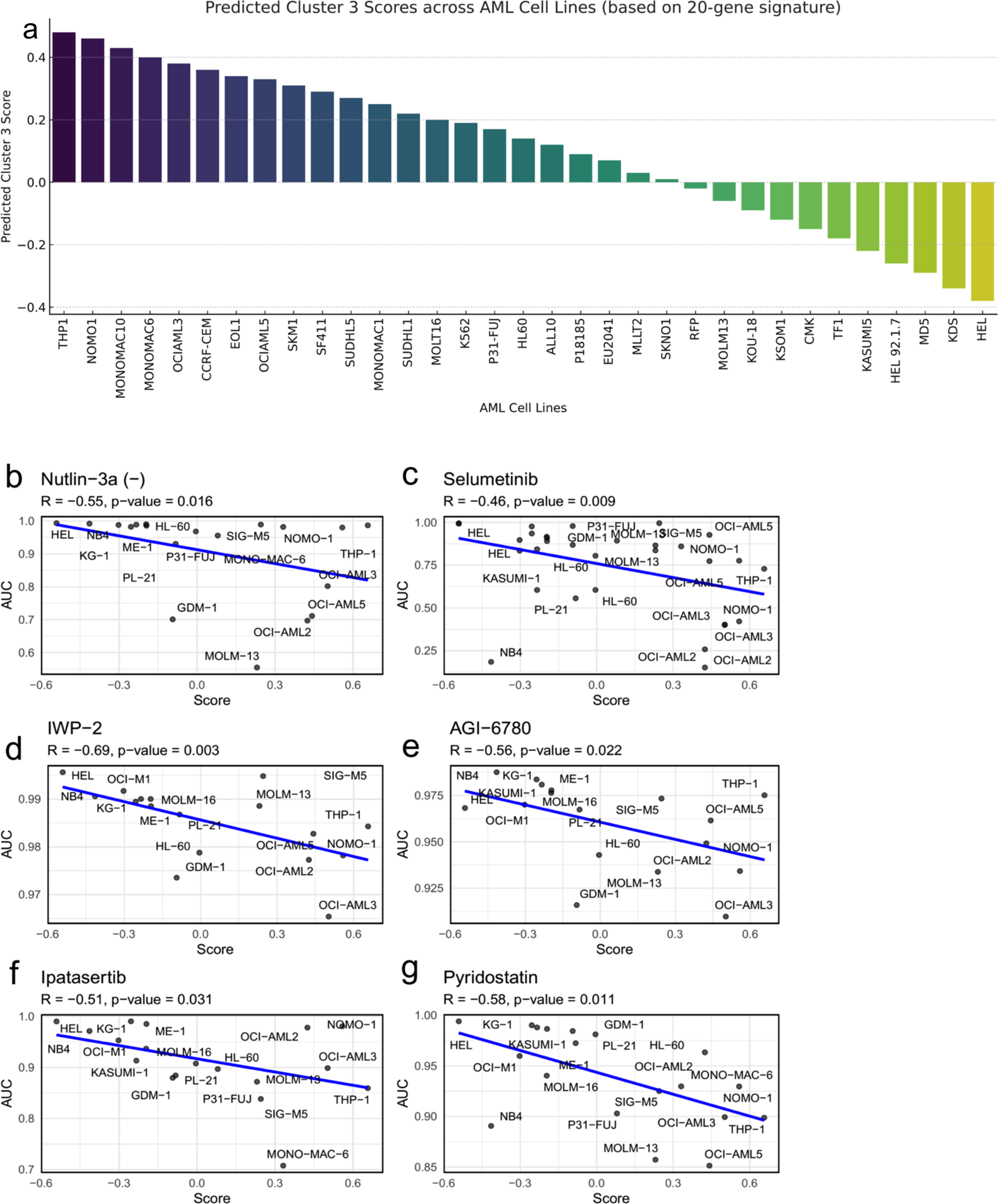
Drug sensitivity analyses in high-risk AML cell lines characterized by a 20-gene signature
To explore the therapeutic potential for targeting high-risk AML subgroups, drug sensitivity was analyzed by correlating IC50 values and AUC metrics from the CTRP with the 20-gene signature scores of AML cell lines (Fig. 5b–g). A significant negative correlation between the 20-gene signature scores and AUC values was used as an indicator of drug sensitivity, where a lower AUC reflected greater efficacy of the drug in inhibiting cell viability. Significant negative correlations were observed between the gene signature scores and AUC for several drugs: Nutlin-3a (-) (R = −0.55, p = 0.016), selumetinib (R = −0.46, p = 0.009), IWP-2 (R = −0.69, p = 0.003), AGI-6780 (R = −0.56, p = 0.022), ipatasertib (R = −0.51, p = 0.031), and pyridostatin (R = −0.58, p = 0.011). These results highlight the effectiveness of the 20-gene signature in identifying high-risk AML cell lines and its potential as a tool for studying drug sensitivities. The observed correlations between gene signature scores and drug efficacy metrics offer valuable insights for exploring novel therapeutic strategies and advancing the development of targeted treatments for high-risk AML.
In vitro experimental validation of the high-risk subgroup under SOC and exploration of new treatment options
To confirm our in silico findings, we aimed to determine whether the 20-gene signature could predict response to SOC chemotherapy treatment. We hypothesized that AML cell lines with high 20-gene signature expression would respond worse to SOC treatment compared with AML cell lines with low 20-gene signature expression. While the effectiveness of SOC in AML depends on a variety of factors (e.g., age, overall health, and specific characteristics of the leukemia), cytarabine was selected for drug response experiments because it represents the backbone of standard induction chemotherapy in AML and remains one of the most widely used and clinically relevant agents in both frontline and salvage settings (Zeidan et al., 2020). To test this hypothesis, we compared the response to cytarabine in cell lines exhibiting a high-risk signature (characterized by a high 20-gene signature score) versus a low-risk signature (characterized by a low 20-gene signature score). Cell lines were ranked based on their relative expression of the 20-gene set (Fig. 5a). We selected MV-411 and THP-1 as representative high-risk group-like cell lines and KG-1 and AML193 as low-risk group-like cell lines. These cell lines were exposed to increasing concentrations of cytarabine to assess sensitivity to this SOC treatment. Our data showed that high-risk group-like cell lines had higher IC50 values (6.8 and 1.9 µM) compared with low-risk group-like cell lines (0.3 and 0.6 µM; Fig. 6a), supporting our hypothesis that high expression of the 20-gene signature is associated with decreased sensitivity to cytarabine.
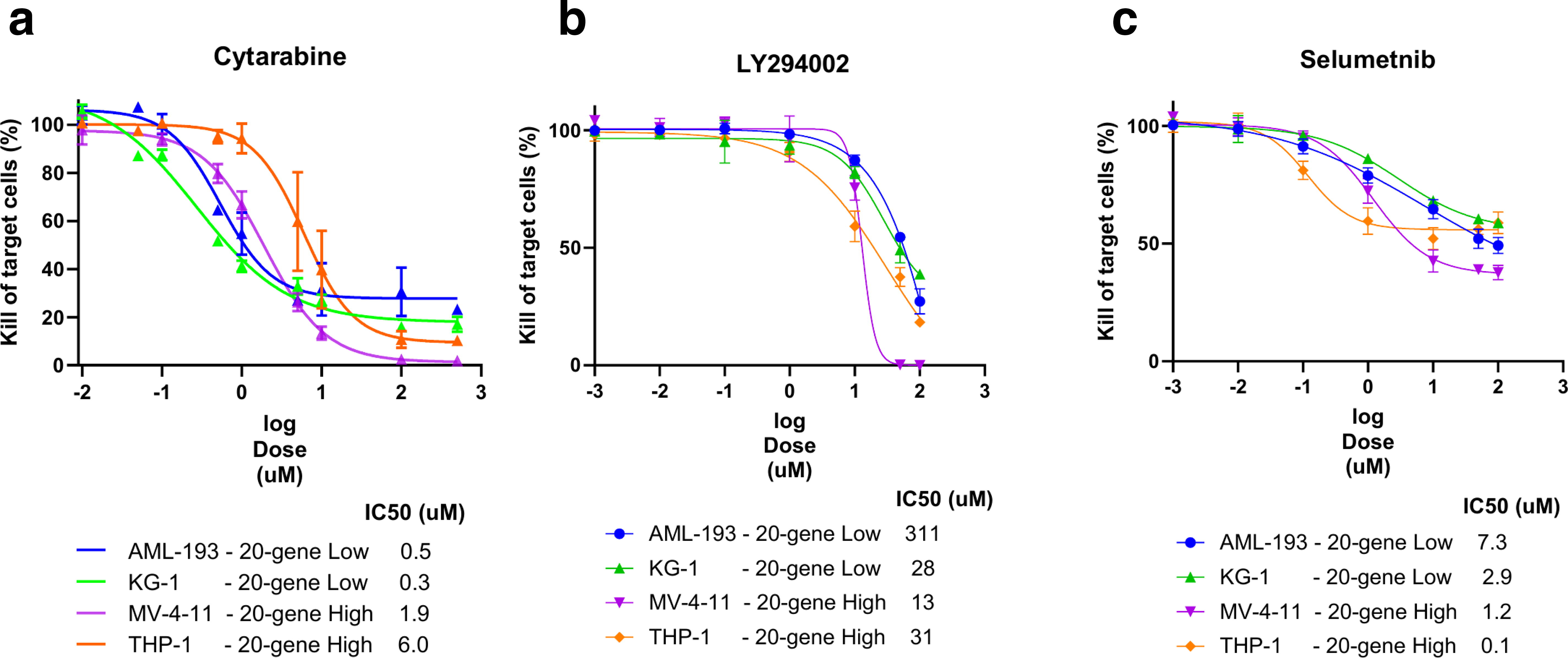
In vitro validation of sensitivity of AML cell lines to cytarabine, PI3K inhibition, and MAPK/ERK inhibition.
To further investigate potential therapeutic strategies for high-risk patients, we used high-risk signature cell lines as a model and explored whether targeting specific transcriptomic features identified in this study could enhance therapeutic options. Based on our transcriptomic analyses, we identified the PI3K and KRAS/MAPK/ERK signaling pathways as potential targets in high-risk patients. These pathways have established roles in cancer and have pharmacological inhibitors available. Both high- and low-risk signature cell lines were treated with a range of concentrations of the PI3K inhibitor LY294002 and the MAPK/ERK inhibitor selumetinib. Our results indicated that high-risk signature cell lines exhibited equal or greater sensitivity to these inhibitors compared with low-risk signature cell lines (Fig. 6b, c, respectively), suggesting that targeting these pathways could be effective in high-risk AML, characterized by the 20-gene signature. These data provide a proof of concept that our molecular profiling can be leveraged to identify novel therapeutic strategies for the treatment of this poor response subpopulation.
Discussion
The integrative analysis conducted in this study highlights the distinct molecular and immune characteristics of a high-risk subgroup of patients with AML within the BeatAML cohort, characterized by poor prognosis. By utilizing NMF on the top 1000 most variable genes, we identified five clusters, with Cluster 3 demonstrating the worst clinical outcome. Genomic profiling revealed that high-risk patients, Cluster 3, harbored significant mutations in FLT3, NPM1, and DNMT3A genes. These mutations are well-documented in AML and are associated with aggressive disease and poor clinical outcomes. FLT3 mutations, particularly internal tandem duplications (ITD), are associated with an aggressive disease course and poor clinical outcomes. FLT3-ITD results in the constitutive activation of the FLT3 receptor, driving uncontrolled cell proliferation and survival (Zhao et al., 2022). NPM1 mutations lead to the aberrant cytoplasmic localization of the NPM protein, disrupting its normal nucleolar functions, including ribosome biogenesis, genomic stability, and regulation of the ARF/p53 tumor suppressor pathway (Rau and Brown, 2009). While NPM1 mutations are often associated with a favorable prognosis in the absence of other high-risk features, their presence in high-risk patients alongside FLT3 and DNMT3A mutations suggests a compounded negative impact on prognosis. DNMT3A mutations, which occur in 20–30% of adult AML cases, are predominantly found in older patients and disrupt epigenetic regulation, influencing hematopoietic stem cell differentiation and self-renewal (Huang et al., 2025). DNMT3A mutation co-occurrence with FLT3-ITD and NPM1 mutations is associated with adverse outcomes in specific patient subsets (Onate et al., 2022).
The activation of oncogenic pathways such as PI3K/AKT/MTOR, RAS/MAPK/ERK signaling in high-risk subgroup further emphasizes its aggressive nature and highlights potential targets for therapeutic intervention. The upregulation of the PI3K/AKT/MTOR pathway, observed in high-risk patients, is a well-known driver of cell growth, proliferation, and survival (Mendez et al., 2019) . Dysregulation of this pathway is common in various cancers, including AML, and is associated with resistance to apoptosis and enhanced survival of malignant cells (Mendez et al., 2019). The RAS/MAPK/ERK pathway is a key signaling cascade in AML, regulating critical cellular processes such as proliferation, survival, and differentiation. Dysregulation of this pathway is frequently implicated in AML progression, contributing to the aggressive behavior of the disease. Its central role in mediating oncogenic signaling makes it an important target for therapeutic interventions, including the use of MEK inhibitors to disrupt downstream signaling and inhibit leukemic cell growth (Berg et al., 2021). The concurrent presence of these genetic alterations and activated oncogenic pathways underlines the aggressive behavior of AML in high-risk patients and points to specific molecular targets for therapeutic intervention. Identifying these key mutations and pathways establishes a molecular framework for the adverse prognosis in this subgroup and supports the development of targeted therapies that could potentially improve clinical outcomes for high-risk patients with AML.
Immune profiling of high-risk subgroup indicated enhanced immune activity, particularly in the complement pathway. The increased presence of M2 macrophages, monocytes, and MSCs suggests an immunosuppressive TME that may contribute to disease progression and resistance to conventional therapies. This complex immune landscape presents both challenges and opportunities for developing immunotherapeutic strategies targeting these immune components.
Validation of our findings in the TCGA-LAML cohort confirmed the robustness and generalizability of the identified high-risk subgroup. The consistent observation of high-risk subgroup in the TCGA cohort indicates the potential clinical relevance of our molecular stratification approach.
The development of a 20-gene signature represents a significant step toward translating the molecular and transcriptomic findings into clinically actionable tools for AML. This signature, derived from the top features identified by the random forest classifier, provides a robust and interpretable framework for stratifying high-risk patients, such as those in Cluster 3, based on their unique molecular profiles. By integrating this signature into our analysis, we demonstrated its ability to reliably distinguish high-risk patients with AML and its utility in identifying cell lines that reflect the aggressive biology of this subgroup.
In vitro validation using AML cell lines from the CCLE provided further insights into the clinical relevance of the 20-gene signature. Cell lines such as THP1 and MV411, which share transcriptomic profiles with high-risk patients with AML, were identified as representative models for this subgroup. These cell lines not only allow for the precise calibration of in vitro assays but also facilitate the evaluation of targeted therapeutic approaches. Moreover, our analysis linked the 20-gene signature with drug sensitivity, revealing that cell lines scoring higher on this signature showed increased sensitivity to several key inhibitors. Notably, drugs such as nutlin-3a (-) and selumetinib exhibited significant negative correlations between gene signature scores and AUC, suggesting that these drugs are particularly effective against cell lines displaying high-risk features. This observation advocates for the use of this 20-gene signature as a biomarker to guide therapy selection in clinical settings, potentially enhancing the precision of treatment strategies deployed against AML.
Conclusions
This study identifies distinct molecular and immune features of a high-risk AML subgroup, with genomic profiling revealing a unique mutation profile in FLT3, NPM1, and DNMT3A. These mutations disrupt critical processes such as signal transduction, genomic stability, and epigenetic regulation, contributing to the aggressive disease phenotype observed in these patients. The concurrent activation of key oncogenic pathways, including PI3K/AKT/MTOR and RAS/MAPK/ERK, further highlights potential molecular vulnerabilities that could be targeted therapeutically.
In addition to these molecular alterations, immune profiling revealed a highly immunosuppressive TME in high-risk patients. Elevated complement activation, increased M2 macrophages, monocytes, and MSCs suggest an immune landscape that contributes to disease progression and resistance to conventional therapies. These findings highlight the importance of integrating immune-targeted strategies with molecular therapies to improve outcomes for these patients.
Further experimental validation showed that cells with genotypes or signatures predicting high-risk patients were less sensitive to the SOC agent cytarabine, as indicated by higher IC50 values, but demonstrated increased sensitivity to targeted therapies, such as PI3K and MAPK/ERK inhibitors, offering promising therapeutic alternatives for this high-risk subtype. Our approach integrates computational analysis of patient datasets with experimental validation to refine AML classification and generate hypotheses for identifying novel personalized medicine strategies.
The development of a 20-gene signature provides a biologically informed tool for stratifying high-risk AML patients and identifying molecular drivers associated with aggressive disease. Validation across transcriptomic datasets and functional studies in AML cell lines further demonstrated its potential in linking genomic and transcriptomic features to drug sensitivity.
We note that biomarker development is a multistage process from analytical validity to clinical validity to clinical and public health utility. The present findings contribute toward a concrete step toward the development of precision/personalized medicine in AML. Future studies in larger samples are necessary, however, to evaluate the present findings. For example, further validation of the 20-gene signature across more diverse global cohorts and exploring the functional implications of identified molecular drivers through in vitro and in vivo studies would be timely. Additionally, investigating the potential of combining genomic and immune profiling with other omics data, such as proteomics and metabolomics, could provide a more comprehensive understanding of AML pathogenesis and lead to the discovery of novel therapeutic targets.
Footnotes
Acknowledgment
The authors thank Inge Verbrugge for her valuable guidance on the in vitro validation assays and for critically reviewing the article.
Authors’ Contributions
K.K., A.C., and H.S. drafted the article. K.K. and A.C. performed the computational analysis and bioinformatics data processing. K.K., H.S., and L.K.B. conceptualized and designed the study. M.G.R. and L.C. conducted the in vitro validation experiments. B.W.H. provided project supervision and guidance. All authors contributed to data interpretation, critical review, and article revisions. All authors read and approved the final article.
Author Disclosure Statement
K.K., M.G.R., L.C., L.K.B., B.W.H., and H.S. are employees of Genmab. The authors declare no other competing interests.
Funding Information
This study was sponsored by Genmab.