
Abstract
Background: The clearance of medical devices by the US Food and Drug Administration (FDA) has remained largely unchanged since 1976, when the Medical Device Amendments Act established a system classifying devices into 3 categories based on safety risk to the consumer. The system allows for the clearance of many orthopedics devices through the 510(k) premarket pathway, which is based on “predicate ancestors,” previously cleared devices that are “substantially equivalent.” Purpose: We sought to trace the predicate ancestors of modern total knee arthroplasty (TKA) devices, specifically those recently cleared for marketing by the 510(k) pathway that claim substantial equivalence to prior devices, despite potential differences in material science and design. In addition, we aimed to document which TKA devices cleared by the 510(k) pathway have substantial equivalence to devices that have since been recalled by the FDA. Methods: To create a comprehensive list of TKA devices, we used FDA Classification Process Codes corresponding to knee arthroplasty to search the FDA’s databases from May 28, 1976, the start of the 510(k) process, to May 1, 2021. Of 1309 resulting devices, 89 were excluded as not related to arthroplasty. For each of the remaining devices, we analyzed the descendant devices that claimed substantial equivalence, either directly or indirectly. We used data of recalled designs to determine both the absolute number of recalled devices and the number of currently cleared devices that presented substantial equivalence claims upon predicates that have since been recalled. Results: Of 1220 knee devices cleared or approved, 6 (0.5%) were approved through the premarket approval application (PMA) process, and 1214 (99.5%) were cleared through the 510(k) pathway. Of the 1214 cleared devices, 217 (17.9%) have been recalled and 204 (16.8%) have ties to at least 1 recalled predicate device linked through generational claims of substantial equivalence. We found 90 devices (7.4%) linked directly to a recalled predicate device. Conclusions: Most knee arthroplasty devices are cleared for marketing through reliance on a complex web of equivalency to previously cleared predicates. We found that many TKA devices thus connected were cleared decades apart, with multiple iterations of design and material modifications. Many currently marketed TKA devices have claimed equivalency to predicates that have been recalled. Our findings suggest the need for novel regulatory strategies that might further patient safety while balancing the unwanted effects of regulatory burden.
Keywords
Introduction
The US Food and Drug Administration (FDA) regulates the clearance and recall of all medical devices in the United States. The pathways to device clearance have remained largely unchanged since 1976 when the Medical Device Amendments Act established a risk-based classification system in which medical devices fall into 1 of 3 classes based on consumer safety risk [9]. Class I devices are low risk with reasonable assurance of safety and effectiveness and do not require premarket approval or clearance. Class II devices are moderate risk and require FDA clearance through a 510(k) premarket submission that demonstrates the device is “substantially equivalent” to a predicate, a previously approved device [10]. Class III devices are high risk and require completion of an FDA premarket approval application (PMA), although some class III devices may be cleared via the 510(k) pathway.
Spending on medical devices increased at an annual rate of more than 6% from 1989 to 2016, contributing $173.1 billion to the national health expenditure [2]. Thousands of companies have taken residence in this packed landscape, with many relying on the streamlined 510(k) process to expeditiously introduce additional devices to the market. The passage of the 21st Century Cures Act of 2016 further reduced the threshold for 510(k) clearance [3,5,6]. In 2017, the FDA cleared 3173 devices, 82% of them through the 510(k) pathway [11].
While class III devices going through the PMA process undergo rigorous and comprehensive review of scientific literature and clinical data prior to final clearance, class II devices going through the 510(k) pathway need not show clinical evidence of equivalence, and fewer than 1% of 510(k) applications do so [8]. Orthopedic devices cleared though 510(k) are recalled at a rate more than 11.5 times higher that of their PMA-approved counterparts [1].
To our knowledge, this is the first study to analyze the predicate devices used to gain clearance of modern knee arthroplasty devices. We aimed to show that recently cleared devices trace their lineage to decades-old ancestral networks of devices claiming substantial equivalence without consideration of potential differences in material science and design. Furthermore, we aimed to document that currently cleared and available devices have equivalency ties to those that have since been recalled by the FDA.
Methods
To create a comprehensive list of knee arthroplasty devices, we used FDA classification process codes that corresponded to knee arthroplasty to search the FDA 510(k) and PMA databases from May 28, 1976 (when the 510(k) process began) to May 1, 2021
Following methods previously described in literature, we mapped out an ancestral network of predicate devices for all 510(k) knee arthroplasty devices cleared between May 1, 2020 and May 1, 2021 (n = 31) [7,12,13]. In our analysis, we examined both the total and the unique number of the predicate devices for each cleared device; we followed the chain of substantial equivalence used to grant clearance to those predicates, as well.
In addition, we analyzed the descendant devices that directly or indirectly claimed substantial equivalence to each of the knee arthroplasty devices cleared from 1976 to 2021. Direct substantial equivalence was determined through review of submitted 510(k) documents in the FDA database [10]. In reviewing these FDA 510(k) documents, we also collected information on each recalled device, including its reason for recall. We analyzed these data to determine the absolute number of recalled devices and to determine the number of descendant devices granted 510(k) approval through claims of substantial equivalence on devices that were later recalled.
We visualized this network of substantial equivalence as bidirectional, with each device as a node involving both forward and backward chronology. Within any given network, the nodes receive connections from predicates (the “parents”) and give connections to devices with substantial equivalence (the “children”). All analyses were conducted in the MATLAB programming language using the “digraph” library.
Results
After excluding unrelated non-arthroplasty items, we completed a list of 1220 knee devices that received FDA approval or clearance from May 28, 1976 to May 1, 2021. Only 6 (0.5%) of these devices were approved through the PMA process, while the overwhelming majority, 1214 (99.5%), were cleared through the 510(k) pathway.
From May 1, 2020, to May 1, 2021, the FDA cleared a total of 31 knee devices through the 510(k) pathway. Of these 31 devices, 1 lacked an FDA summary form with 510(k) predicate information. The remaining 30 devices claimed an average number of 3.1 direct predicates, defined as the devices directly cited within the summary form as substantially equivalent (range: 1–8). After removing duplicate predicates, we found a total of 78 unique direct predicates with clearance dates between 1996 and 2021, averaging 7.1 years before the end of our study period of May 2021. Furthermore, in the same time period, 4 of the 30 devices (12.9%) claimed direct substantial equivalence to devices recalled due to design, while an additional 21 out of 30 (70%) claimed indirect substantial equivalence to devices recalled due to design.
We investigated the direct and indirect predicates of the 30 cleared devices, and we found significant overlap in their predicate networks. We found significant overlap in their ancestral networks of substantial equivalence. Of the 30 cleared devices, 24 were categorized as knee systems, 5 as femoral components, and 1 as a liner. In addition, these 30 devices cited substantial equivalence with a total of 1301 510(k) approved predicate devices. When duplicates were removed, a total of 409 unique predicates remained. The number of ancestral predicates ranged from 1 to 101 (mean = 43.4)
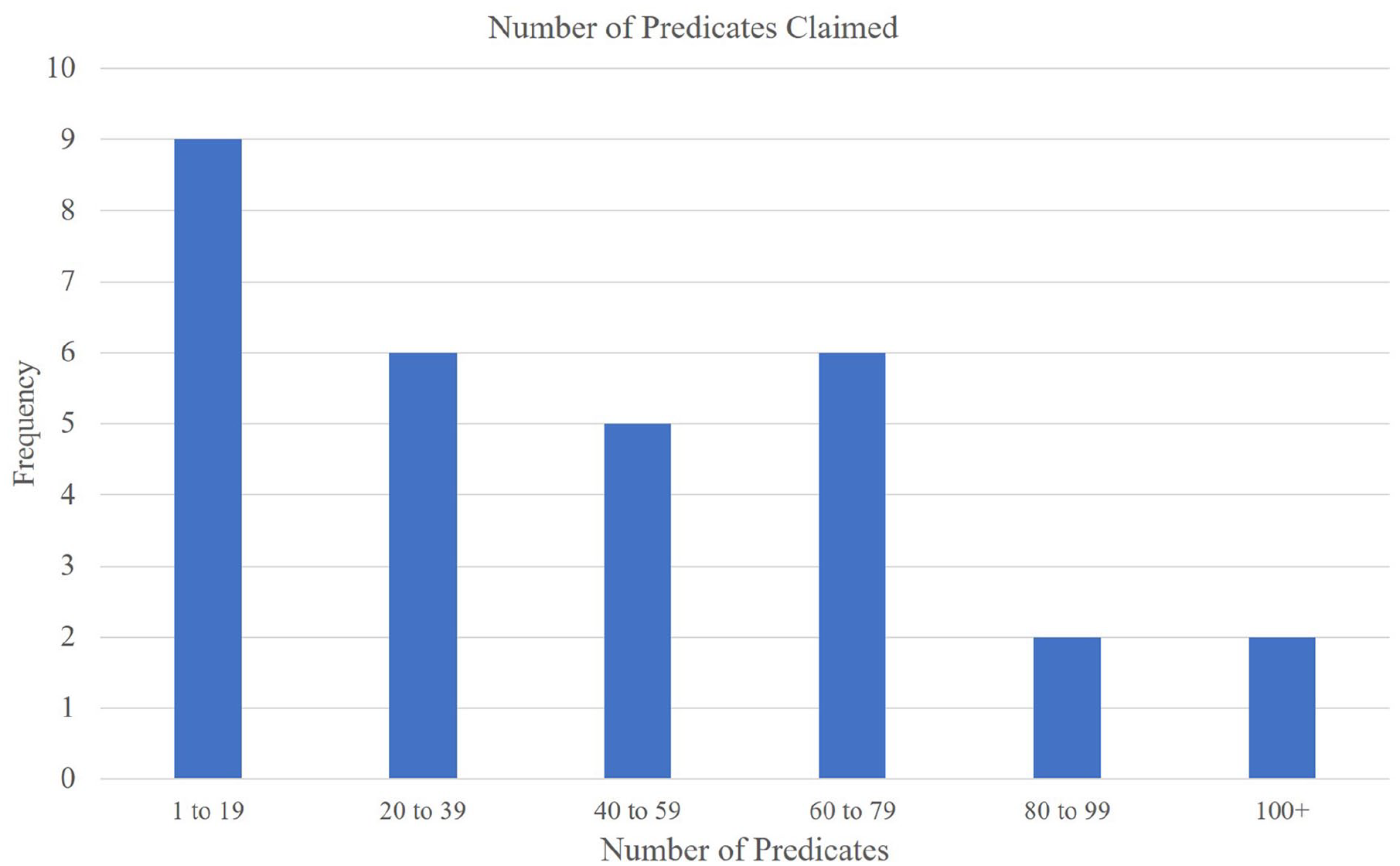
Predicates claimed (directly and indirectly) for each knee arthroplasty device cleared between May 1, 2020 and May 1, 2021 (n = 30).
Overall, the 30 cleared devices claimed substantial equivalents from 1984 to 2021, and the visual network of ancestry reveals highly interwoven networks linking the devices together (Fig. 2). The true substantial equivalence graph is likely even more complex, because while 633/868 (72.9%) of devices approved through the 510(k) pathway from April 1996 to May 2021 provided predicate information, none of the 349 devices approved through the 510(k) pathway before April 1996 provided this information, as there was no summary data found in the FDA online database.
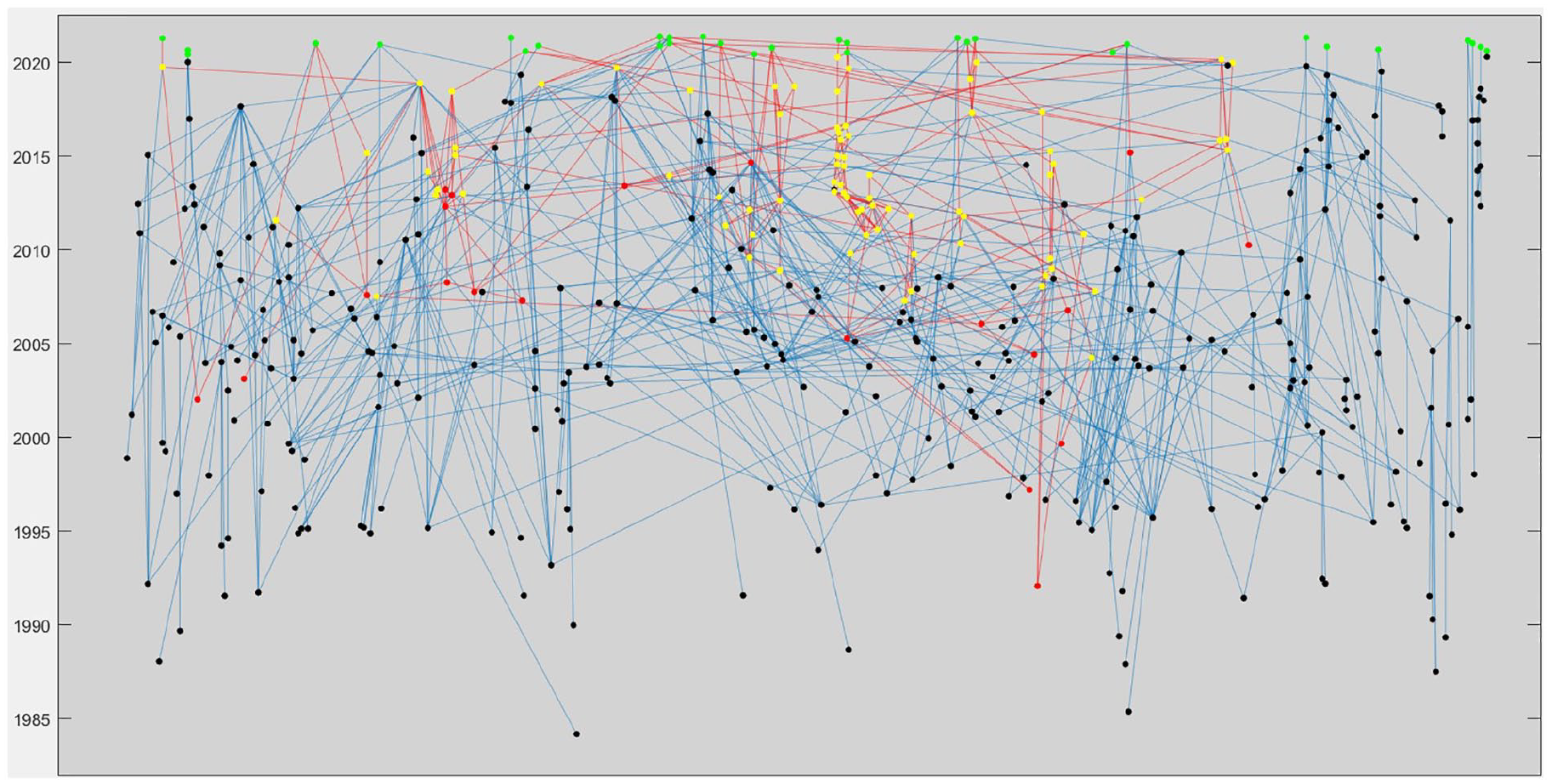
Ancestral graph of devices cleared between May 1, 2020,- and May 1, 2021. Each node represents a device, and lines between nodes represent claims of substantial equivalences. The Y-axis represents the year each device was cleared. Green = devices cleared between May 1, 2020 and May 1, 2021 Yellow = devices descended from a device recalled for design issues Red = devices that have been recalled; devices connected via a red line have claimed at least one predicate that has been recalled.
Examining the equivalence network at the level of categorization, we found that of the 409 unique predicates there were 30 recently cleared devices declared substantially equivalent. Of these, 302 (73.8%) were knee implant systems, 48 (11.8%) were tibial components, 31 (7.6%) were femoral components, 17 (4.2%) were liners, and 11 (2.7%) were patellar components (
The total number of predicates associated with each of the 30 recently cleared devices can exceed the number of devices directly associated in each category due to interconnected links in the network of ancestry.

Upon examination of knee arthroplasty devices cleared from 1976 to 2021, we found a range of descendants from 0 to 146. Two of the 6 devices approved through PMA during this time had descendants, while 657 out of 1220 (53.9%) devices cleared through 510(k) had descendants that claimed substantial equivalence. The remaining 563 510(k) devices had no descendants (Fig. 3). The 657 510(k) devices with substantial equivalence claims each had an average of 14.2 descendants. Of these, 9 of the devices that were released from 1992 to 2002 had more than 100 descendant devices.
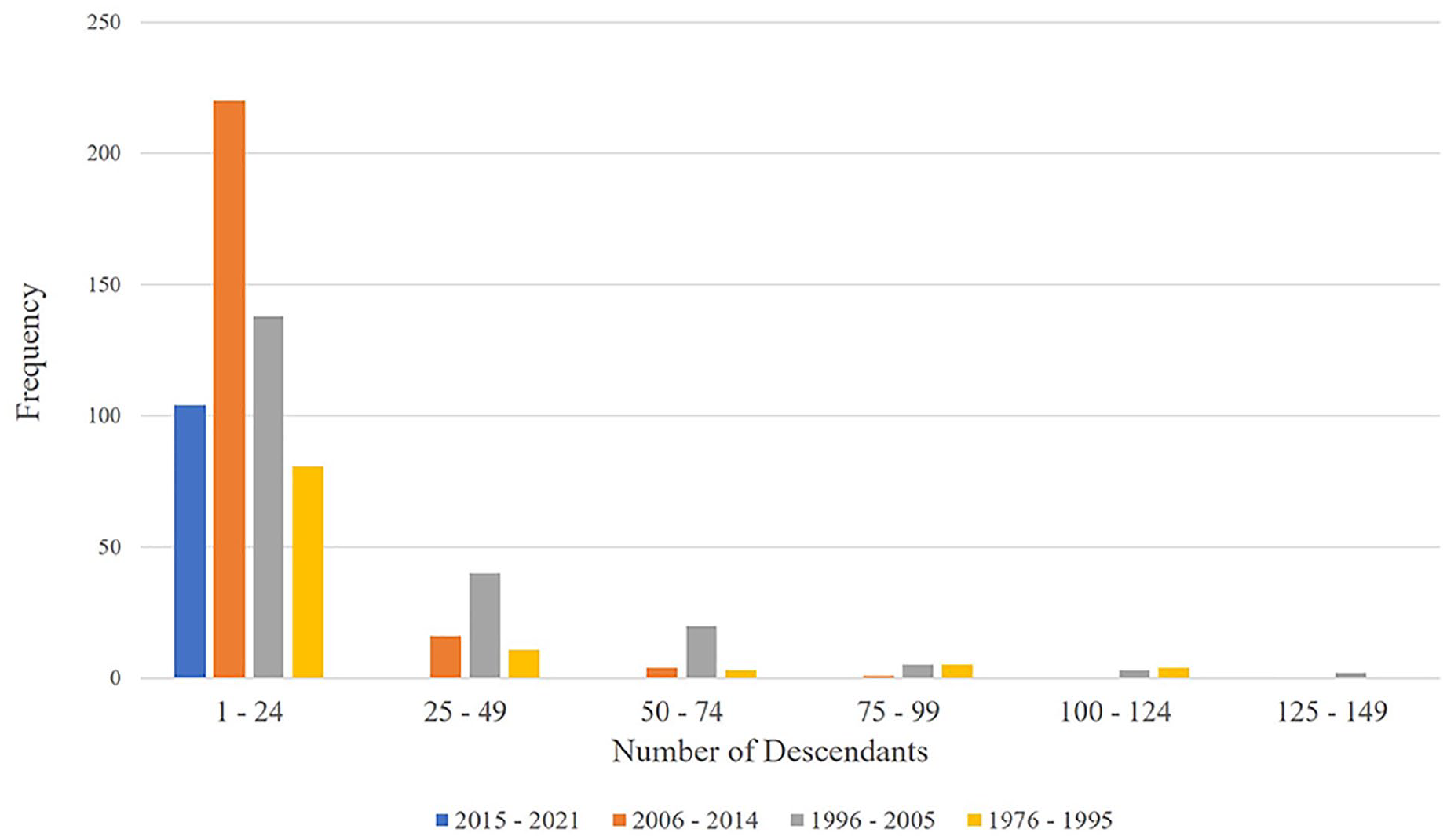
The total number of descendants associated with each ancestral 510(k) predicate across all generations of substantial equivalence. Red = recalled, yellow = direct predicate was recalled, green = not recalled and does not have a direct predicate that was recalled. Key with identification of each numbered implant included in Supplement 4. Device 1 represents the recalled device K052917 that served as predicate for 78 devices. Devices with zero descendants (n = 563) were excluded from this figure.
Of the 1214 knee arthroplasty devices that have passed through the 510(k) pathway since 1976, 217 (17.9%) have been recalled for several reasons, including 72 (33.2%) that were recalled for issues with quality control, 46 (21.1%) for packaging, 37 (17.1%) for labeling, 30 (13.8%) for implant-replated design, 24 (11.1%) for non-implant-related design, 7 (3.2%) for storage, and 1 (0.5%) for manufacturing. Our researched focused on the subgroup of 30 devices that were recalled due to deficiencies in implant design. Of these, 2 were for class III devices, indicating recalls of products that could cause serious health problems or death, while 28 were for class II devices, indicating recalls of products that might cause serious injury or temporary illness. Most of the design recalls, 24 (80.0%), were for devices classified as implant systems, followed by design recalls of 5 (16.7%) for tibial components, and 1 (3.3%) for femoral components
In all, of the 1214 knee arthroplasty devices evaluated, 204 (16.8%) have ties to at least 1 recalled predicate device linked through generational claims of substantial equivalence, with 90 devices (7.4%) linked directly to a recalled predicate device. One device (K052917: Stryker Compartmental Knee System) was recalled due to increased rate of revision surgery; it served as a predicate to 78 descendant devices
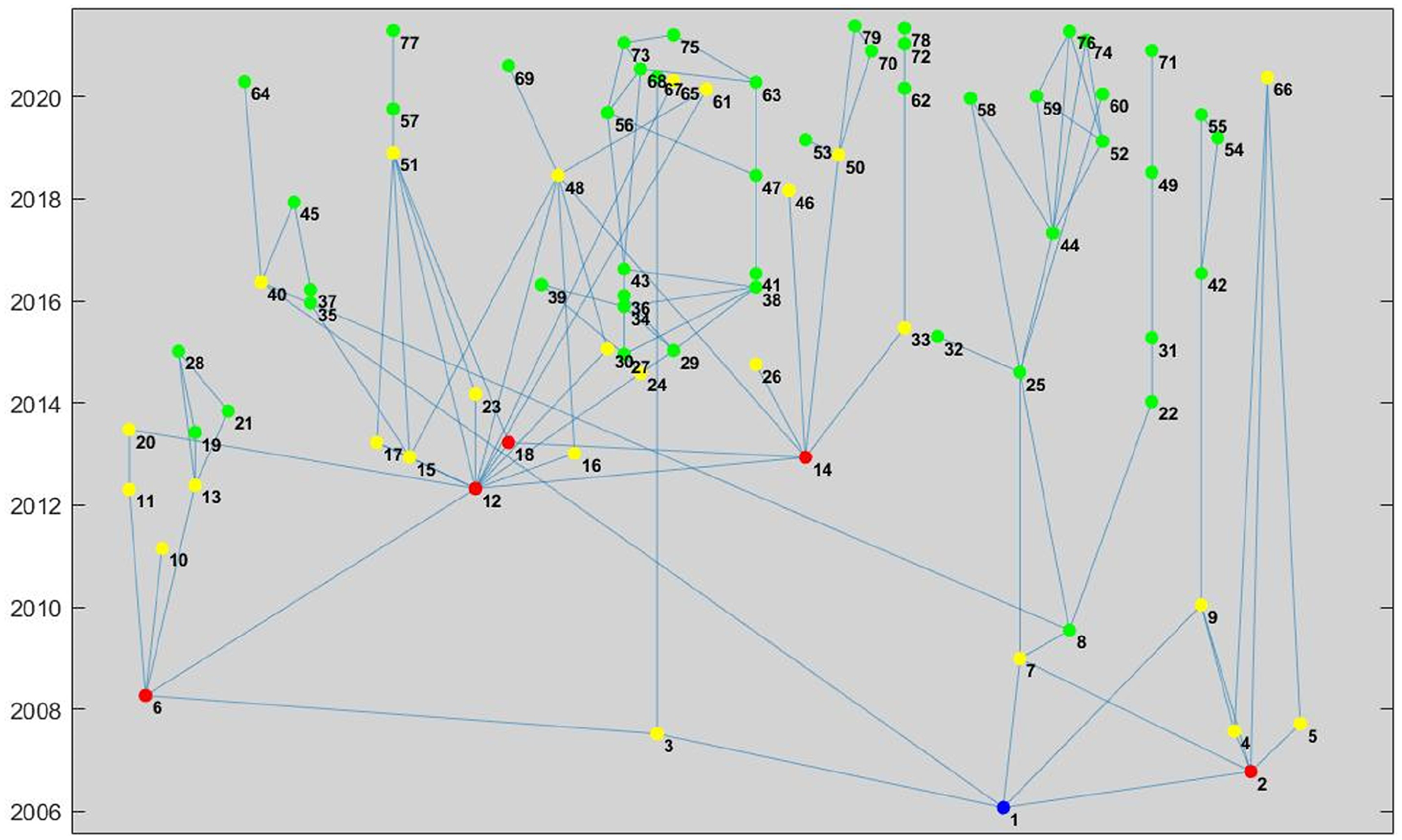
All descendants from 1 recalled device (K052917) that was cleared in 2005.
Discussion
The United States already has the highest incidence rate of knee arthroplasty in the world, with contributing factors that include an aging population and rising rates of obesity and osteoarthritis. By 2030, the use of knee arthroplasty is projected to increase by 182% [4], potentially placing pressure on manufacturers to produce new and improved knee prostheses that require clearance through either the PMA or 510(k) pathway. While the majority of knee implants have been used with excellent success rates and survivorship, over the years specific knee devices have been pulled from the market for concerns over design and safety. In this study, we reviewed the 510(k) clearance process and traced the ancestry of predicate devices upon which nearly all knee arthroplasty implants have been cleared since the inception of the Medical Device Amendment Act in 1976. Only 6 class II devices, all mobile-bearing devices, have been approved through the rigorous PMA process, while the rest have all been cleared through the 510(k) process, including all knee implants cleared during a recent 2-year period (May 1, 2018, to May 1, 2020).
There were several limitations to this study. First, we identified devices by their FDA clearance date, but some cleared devices may never have made it to market. Second, we reviewed the list of knee arthroplasty devices and removed those that were clearly unrelated (eg, bone cement), and it is possible that we missed certain devices in this manual process. In addition, we classified devices into categories such as implant system and femoral, patellar, and tibial components, and it is possible that devices were miscategorized in this process. Fourth, our analysis treated each 510(k)/PMA summary as a unique device, but some devices may have had multiple applications submitted over time. Fifth, this study used the FDA database to find predicate information. While the majority of implants since 1996 provided summaries, very few implants before 1996 contained summary information. This limited our evaluation of older devices, and devices cleared before 1996 would likely provide a few more generations of predicates. Thus, our study likely underestimated the connectedness of knee arthroplasty devices, and more devices from pre-1996 are likely to serve as predicates. Finally, design recall data were used as a benchmark for design flaws, but it is possible that many design recalls were due to minor issues that the manufacturer rectified.
While each 510(k) cleared device cites only a few direct predicates, a mapping of the ancestral network of knee implants reveals a large number of predicates. Ultimately, this process creates a network of devices that transitively claims to be “substantially equivalent,” despite being cleared decades apart. As presented in
Furthermore, some currently marketed devices descended from predicates that have been recalled due to design issues. Fig. 4 highlights a device recalled for design issues, with ties to dozens of devices on the market, including 12 of the devices approved from May 2020 to May 2021. A 2011 Institute of Medicine report suggested that new strategies should be implemented to improve upon the 510(k) process and ensure the safety and efficacy of medical devices. Prior attempts to address shortcomings in the 510k clearance process, including the Safety of Untested and New Devices Act of 2012, were ultimately not passed into law [4].
For more than 4 decades, the 510(k) process has provided a rapid and efficient path for medical devices to come to market and enabled patients to benefit from these devices in a relatively expeditious manner. By requiring high-volume clinical testing prior to clearance of all implants, the regulatory burden and cost of bringing new devices to market would increase, which could have significant downstream effects such as an increase in health care spending. Policymakers have argued that to balance cost and efficiency, strategies are needed that limit the regulatory burden and also evaluate device safety prior to widespread use.
In conclusion, knee arthroplasty devices exhibit a complex web of equivalence through claims of predicates. However, many connected devices were cleared decades apart with multiple iterations of design and material modifications. Furthermore, many currently marketed knee arthroplasty devices have claimed equivalence to predicates that have subsequently been recalled. Novel regulatory strategies should be explored.
Supplemental Material
sj-docx-1-hss-10.1177_15563316221099014 – Supplemental material for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway
Supplemental material, sj-docx-1-hss-10.1177_15563316221099014 for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway by Andrew Zhu, Xiaohan Ying, Christian A. Pean, Neil P. Sheth, Michael B. Cross, Alejandro Gonzalez Della Valle and Ajay Premkumar in HSS Journal®: The Musculoskeletal Journal of Hospital for Special Surgery
Supplemental Material
sj-docx-2-hss-10.1177_15563316221099014 – Supplemental material for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway
Supplemental material, sj-docx-2-hss-10.1177_15563316221099014 for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway by Andrew Zhu, Xiaohan Ying, Christian A. Pean, Neil P. Sheth, Michael B. Cross, Alejandro Gonzalez Della Valle and Ajay Premkumar in HSS Journal®: The Musculoskeletal Journal of Hospital for Special Surgery
Supplemental Material
sj-docx-3-hss-10.1177_15563316221099014 – Supplemental material for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway
Supplemental material, sj-docx-3-hss-10.1177_15563316221099014 for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway by Andrew Zhu, Xiaohan Ying, Christian A. Pean, Neil P. Sheth, Michael B. Cross, Alejandro Gonzalez Della Valle and Ajay Premkumar in HSS Journal®: The Musculoskeletal Journal of Hospital for Special Surgery
Supplemental Material
sj-docx-4-hss-10.1177_15563316221099014 – Supplemental material for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway
Supplemental material, sj-docx-4-hss-10.1177_15563316221099014 for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway by Andrew Zhu, Xiaohan Ying, Christian A. Pean, Neil P. Sheth, Michael B. Cross, Alejandro Gonzalez Della Valle and Ajay Premkumar in HSS Journal®: The Musculoskeletal Journal of Hospital for Special Surgery
Supplemental Material
sj-docx-5-hss-10.1177_15563316221099014 – Supplemental material for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway
Supplemental material, sj-docx-5-hss-10.1177_15563316221099014 for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway by Andrew Zhu, Xiaohan Ying, Christian A. Pean, Neil P. Sheth, Michael B. Cross, Alejandro Gonzalez Della Valle and Ajay Premkumar in HSS Journal®: The Musculoskeletal Journal of Hospital for Special Surgery
Supplemental Material
sj-docx-6-hss-10.1177_15563316221099014 – Supplemental material for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway
Supplemental material, sj-docx-6-hss-10.1177_15563316221099014 for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway by Andrew Zhu, Xiaohan Ying, Christian A. Pean, Neil P. Sheth, Michael B. Cross, Alejandro Gonzalez Della Valle and Ajay Premkumar in HSS Journal®: The Musculoskeletal Journal of Hospital for Special Surgery
Supplemental Material
sj-docx-7-hss-10.1177_15563316221099014 – Supplemental material for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway
Supplemental material, sj-docx-7-hss-10.1177_15563316221099014 for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway by Andrew Zhu, Xiaohan Ying, Christian A. Pean, Neil P. Sheth, Michael B. Cross, Alejandro Gonzalez Della Valle and Ajay Premkumar in HSS Journal®: The Musculoskeletal Journal of Hospital for Special Surgery
Supplemental Material
sj-xlsx-8-hss-10.1177_15563316221099014 – Supplemental material for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway
Supplemental material, sj-xlsx-8-hss-10.1177_15563316221099014 for The Complex Process of Using the Interconnected Knee Arthroplasty Device Clearance Pathway by Andrew Zhu, Xiaohan Ying, Christian A. Pean, Neil P. Sheth, Michael B. Cross, Alejandro Gonzalez Della Valle and Ajay Premkumar in HSS Journal®: The Musculoskeletal Journal of Hospital for Special Surgery
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Neil P. Sheth, MD, has relationships with Medacta, Microport, Smith & Nephew, Zimmer, Elsevier, AAOS and the Eastern Orthopaedic Association. Michael Cross, MD, has relationships with 3M, DePuy A Johnson & Johnson Company, Exactech, Flexion Therapeutics, Intellijoint, Smith & Nephew, BICMD, Imagen, Insight Medical, Parvizi Surgical Innovation, Flexion Therapeutics, Bone and Joint Journal 360, the Journal of Orthopaedics and Traumatology, and Techniques in Orthopaedics. Alejandro Gonzalez Della Valle, MD, has relationships with Johnson & Johnson, Link Orthopaedics, Naviswiss, OrthoDevelopment, and OrthoSensor. Andrew Zhu, BSE, Xiaohan Ying, BS, Christian A. Pean, MD, MS, and Ajay Premkumar, MD, MPH, declared no potential conflicts of interest.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Human/Animal Rights
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2013.
Informed Consent
Informed consent was not required for this study.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article as supplemental material.
CME Credit
Level of Evidence
Level V: Database Study.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.