
Abstract
Hematopoietic stem cells (HSCs) are multipotent stem cells capable of differentiating into all types of blood and immune progeny and endowed with self-renewal capacity to enable lifelong hematopoiesis. Based on these unique characteristics, HSCs are utilized as a cell source for cell and gene therapies. During its dawning period, HSC-based therapies faced significant challenges due to inefficient gene transfer and insertional leukemogenesis. However, technological advances, such as the use of HIV-derived lentiviral vectors and cellular promoters, have established HSC gene therapy as a powerful treatment modality for patients with congenital monogenic diseases, leading to approved therapies commercially available in Europe and the United States. HSC-based therapies are now being explored for broader indications, including cancer, autoimmune, and infectious diseases. Innovative concepts achievable with HSCs—such as delivering therapeutic proteins to hard-to-reach tissues, in vivo delivery of antibodies and immune cells, and molecular shielding—have been proposed, offering new therapeutic approaches. Moreover, technological innovations in related fields, including more precise gene expression control and reduced-toxicity bone marrow conditioning, are expanding the range of applications. HSC-based cell and gene therapies are therefore evolving into a therapeutic modality applicable beyond monogenic diseases to a broader range of indications, to provide therapeutic value to patients with intractable diseases.
Introduction
Hematopoietic stem cells (HSCs) are multipotent, somatic stem cells able to differentiate into all types of blood and immune cells, endowed with self-renewal capacity to sustain the hematopoietic system throughout life. 1 HSC transplantation (HSCT) is established as a curative treatment for many hematological disorders, including malignancies. Given these characteristics and the well-established transplantation technology, HSCs are considered one of the most promising target cells for cell and gene therapy. In recent years, protocols have been established to treat congenital disorders caused by monogenic mutations by transplanting HSCs genetically modified outside the patient’s body. Key characteristics of HSCs, such as their long-term persistence and ability to differentiate into diverse cell types, hold the potential to overcome limitations of conventional cell therapies using terminally differentiated cells (adoptive transfer). Therefore, the potential applications of HSC-based cell and gene therapy are not limited to monogenic diseases. 2 Active research is underway to apply HSC gene therapy to broader applications, including cancer and autoimmune diseases. This short review introduces the history, current achievements, latest initiatives, and key technologies leading to future expansion in this rapidly advancing field.
Hematopoietic Stem Cells and Hematopoietic Stem Cell Transplantation
The roots of HSC research in mammals lie in experiments conducted in the 1950s, where mice exposed to radiation were rescued through bone marrow transplantation. Lorenz et al. reported the existence of cells within the mouse bone marrow possessing the ability to reconstruct the damaged hematopoietic system, providing the first functional evidence of the existence of HSCs. 3 Subsequently, the development of colony-forming assays and flow cytometry enabled the analysis of cell surface molecular phenotypes and the differentiation potential of individual cells at the single-cell level.4–6 This allowed the identification of cells possessing multipotency as tangible entities and revealed the hierarchy of the hematopoietic system with HSCs at its top.
Mammalian HSCs originate from the mesoderm, developing from vascular endothelial cells in the fetal Aorta-Gonad-Mesonephros region. They subsequently migrate to the fetal liver and, in the perinatal period, to the bone marrow. It has been established that adult HSCs in both primates and rodents reside primarily in the bone marrow. 7
Human HSCs can be analyzed and isolated using flow cytometry techniques based on specific cell surface markers. It is known that the combined use of multiple positive markers [e.g., cluster of differentiation (CD) 34, CD90] and negative markers [e.g., lineage marker, CD38, CD45RA] allows for highly enriched isolation of human HSCs. However, a truly selective marker defining HSCs has not yet been identified, necessitating retrospective confirmation using functional assays such as colony formation or transplantation into immunodeficient animals. Generally, isolated CD34-positive hematopoietic stem/progenitor cells (HSPCs) are used for transplantation.7–9
HSCT is a procedure that involves transplanting HSCs from the recipient’s own body (autologous transplantation) or from a donor (allogeneic transplantation) to the recipient to reconstitute the hematopoietic and immune systems. HSCT is used to restore a hematopoietic system irreversibly damaged by intense chemotherapy or radiation therapy, or to eliminate cancer cells via the graft-versus-leukemia/lymphoma activity of allogeneic grafts. The clinical practice of HSCT originated from pioneering research on allogeneic bone marrow transplantation led in the 1950s by E Donnall Thomas.10,11 Subsequent improvements in pre-transplant conditioning regimen, overcoming HLA compatibility issues, and managing Graft-versus-Host Disease (GvHD), have established HSCT as a curative treatment for refractory hematological disorders and blood cancers.12,13 The major therapeutic sources of HSCs are bone marrow, peripheral blood, and umbilical cord blood. In adults, hematopoietic stem cells are primarily found in the bone marrow. It is known that HSCs stimulated by mobilizing agents such as Granulocyte-Colony Stimulating Factor (G-CSF) or C-X-C motif chemokine receptor 4 (CXCR4) antagonists can be mobilized into the peripheral blood. This has been established as a relatively less invasive method for collecting HSCs. 14
Genetic Modification Technology for HSCs and Its Clinical Applications
Genetic modification technology for HSCs offers significant hope for a cure for congenital monogenic diseases difficult to treat by conventional therapeutic modalities, revolutionizing the treatment paradigm. The foundation for this approach was laid in the 1960s and 1970s, and it continues to develop despite facing challenges in clinical application.
The biological characteristics of HSCs make them an ideal target for gene therapy. The effects of genetic modification in these cells may extend to all types of blood and immune cells throughout the body and persist throughout the patient’s lifetime. This offers patients the possibility of “cure with a one-time treatment”. The foundation of current gene therapy was shaped by the development of techniques for introducing exogenous genes into mammalian cells using viral vectors, which began in the 1960s, and by the vigorous discussions surrounding the application of recombinant DNA technology in the 1970s.15–17 In 1984, the introduction of an exogenous gene into murine HSCs using a γ retroviral vector was reported, demonstrating that the genetically modified HSCs maintained their stem cell properties. 18 The earliest application to human disease treatment began with the therapy for adenosine deaminase-deficient severe combined immunodeficiency (ADA-SCID) conducted in the early 1990s. These studies demonstrated that human HSCs supplemented with a normal copy of the adenosine deaminase gene could produce functionally normal T cells in the human body, showing the power of HSC gene therapy.19,20 Later in the 1990s and 2000s, clinical trials for X-linked severe combined immunodeficiency (SCID-X1), ADA-SCID, and Wiskott-Aldrich syndrome (WAS) proved the efficacy of HSC gene therapy in replacing a defective immune system.21–23 Unfortunately and unexpectedly, six of the 20 patients treated for SCID-X1 and 7 of the 10 patients treated for WAS developed leukemia within a few years after treatment.24–27 These serious adverse events had a major impact on the development of gene therapy. The cause of leukemogenesis was the insertion of the γ-retroviral vector used to deliver the normal gene copy in a nearby proto-oncogene, notably LIM domain only 2 (LMO-2), which caused its transcriptional activation and deregulation. 28
To overcome the apparent strong genotoxicity of γ-retroviral vectors, lentiviral vectors were developed by modifying the human immunodeficiency virus-1 (HIV-1). Lentiviral vectors efficiently transduce non-proliferating cells, enabling high-efficiency gene delivery into quiescent HSCs without cytokine stimulation.29–31 Crucially, they prevent activation of neighboring genes through a modification called Self-Inactivation (SIN), which removes the U3 enhancer/promoter region from the long terminal repeat (LTR). This mechanism inactivates LTR-driven transcription while preventing activation of neighboring genes, significantly reducing the risk of leukemogenesis due to insertional mutations. 29 The currently mainstream, third-generation lentiviral vectors prevent the production of self-replicating viruses by encoding essential viral components (gag, pol, env, etc.) in separate plasmids. They reduce toxicity and immunogenicity by deleting accessory genes from the HIV-1 genome. Furthermore, it has been reported that γ-retroviruses and lentiviruses differ in the components of the Pre-Integration Complex (PIC) formed after cellular entry, leading to distinct patterns of insertion in the genome. Compared to γ-retroviruses, which insert near histone-acetylated promoter and enhancer regions, lentiviruses integrate primarily in intronic regions of active genes away from regulatory regions, further contributing to their higher safety profile.28,32–34 These advances have evolved lentiviral vectors into a safer technology for clinical applications, and are currently used in all HSC gene replacement therapies.
HSC Gene Therapy for Congenital Monogenic Diseases
After a period of stagnation due to the safety issues encountered in early clinical trials, HSC gene therapy saw renewed progress in the 2010s thanks to numerous efforts of scientists in this field. Technological improvements included the development of safer gene delivery vectors, precise gene expression control using improved promoters and enhancers, and the advancement of genome editing technology, exemplified by the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated proteins (Cas) system, enabling highly efficient and accurate genome editing. In recent years, at least five ex vivo autologous HSC gene therapies have gained regulatory approval for diseases caused by monogenic mutations, such as cerebral adrenoleukodystrophy (C-ALD), ß-thalassemia, sickle cell disease, metachromatic leukodystrophy (MLD), and WAS.35–41 HSC gene therapy consists of three fundamental steps: (1) collection of HSCs from the patient, (2) ex vivo gene introduction/modification, and (3) transplantation back into the patient. These novel treatments demonstrate dramatic and sustained effects for diseases that previously lacked effective therapies, revolutionizing the treatment paradigm. Simultaneously, they raise issues for broader societal adoption, such as the reimbursement of high treatment costs. 42 This is a very important topic when considering the sustainability and future expansion of this technology, but it is beyond the scope of this short review.
HSC-Based Cell and Gene Therapy: Beyond Monogenic Diseases
The success of HSC gene therapy in rare congenital diseases suggests the potential application of this established principle and technology to a broader range of disorders. Its potential scope is extensive, with active exploration of applications in cancer, autoimmune diseases, central nervous system disorders, metabolic diseases, and even infectious diseases like AIDS. (See Fig. 1)
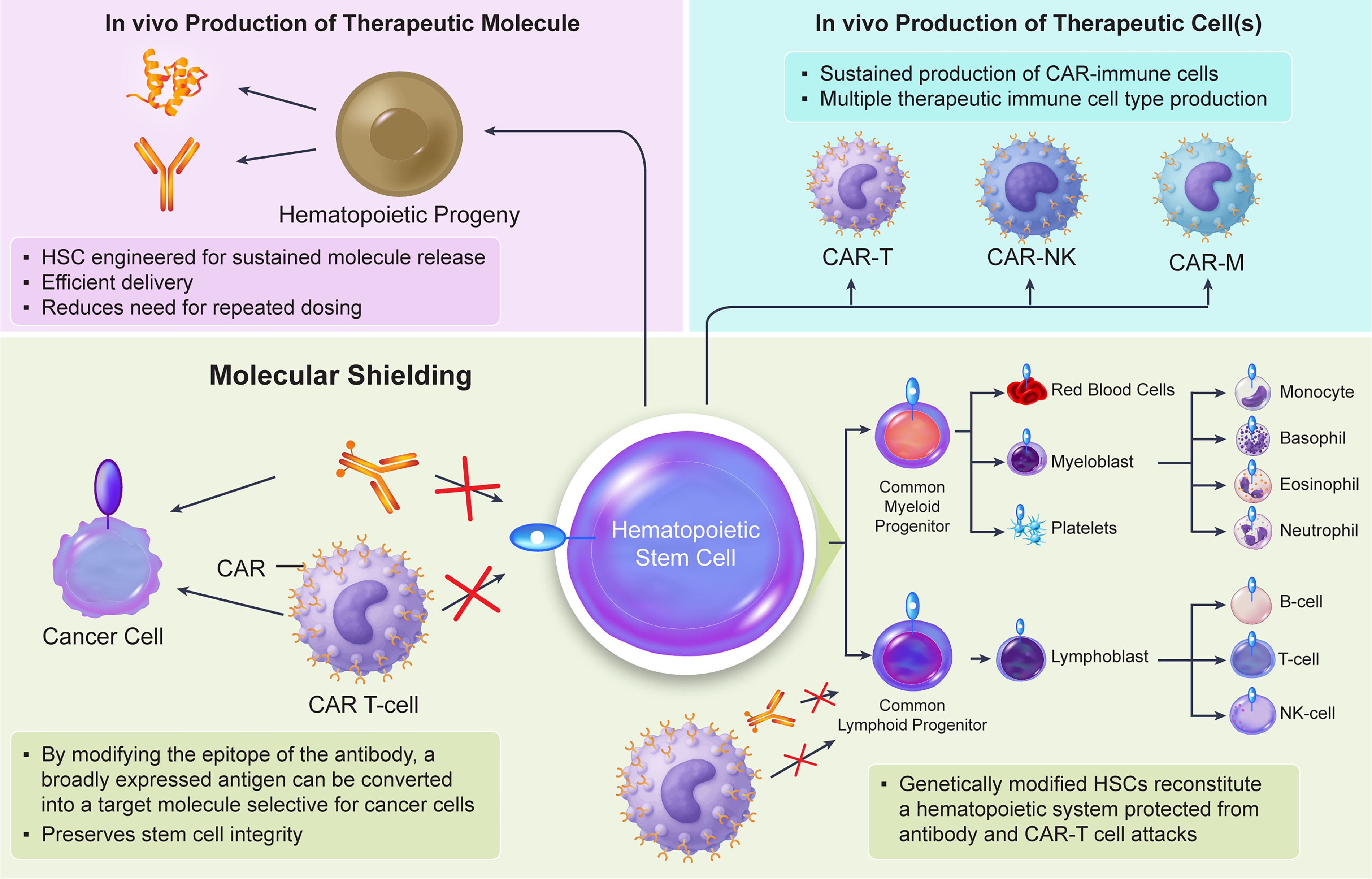
Key emerging concepts realizable with genetically engineered HSCs.
An example of the “beyond monogenic” domain is Crohn’s disease, an inflammatory bowel disorder characterized by chronic inflammation of the intestinal mucosa. While anti-inflammatory drugs and surgical treatments are the current standard of care, no curative treatment exists. It is known that 20%–40% of Crohn’s disease patients carry a mutation or a single nucleotide porimorphism (SNP) in nucleotide-binding oligomerization domain-containing protein 2 (NOD2) gene, encoding a molecule that recognizes bacterial peptidoglycans. NOD2 is expressed in monocytes, macrophages, and Paneth cells, and acts as a sensor of bacterial invasion. A reduction in NOD2 function decreases the innate immune system’s ability to recognize pathogens in the intestine, leading to excessive inflammation driven by the adaptive immune system. Consequently, mutations in this gene may contribute to the development of Crohn’s disease, while patients with biallelic NOD2 mutations exhibit an early-onset, severe, stricturing disease refractory to conventional treatment.43–45 Transplantation of autologous HSCs supplemented with a normal copy of the NOD2 gene in NOD2-mutated patients is expected to restore the patient’s normal pathogen recognition in the macrophage progeny, potentially improving or even curing the disease. After transplantation, monocytes generated by genetically modified HSCs migrate to the intestinal epithelium and differentiate into macrophages, a key component of the intestinal innate immune system. This approach is expected to become a new treatment for refractory diseases that are difficult to cure. Enjalbert et al. recently showed that restoring the normal NOD2 gene by lentiviral vector transduction into lineage marker negative HSPCs from Crohn’s disease patients harboring biallelic NOD2 mutations restores innate immunity responses to muramyl dipeptide, a bacterial cell wall component, confirming the promise of this approach.46,47
Immune cells differentiated from HSCs are distributed throughout body tissues. This includes tissues difficult for drugs to reach, such as the brain and bone. By incorporating genes encoding therapeutic proteins into HSCs, it becomes possible to deliver these proteins to target tissues via their immune cell progeny such as tissue macrophages. This concept has already been clinically validated in lysosomal storage disorders; for MLD, Atidarsagene autotemcel achieves delivery of the normal arylsulfatase A gene and protein to the patient’s brain, with demonstrated clinical efficacy.
40
Similarly, in patients affected by mucopolysaccharidosis type I, Hurler variant, the progeny of autologous HSCs supplemented with a wild-type alpha-
A pioneering effort applying this concept to solid tumors is TemferonTM, developed by Genenta Science. Temferon is an autologous CD34 + HSPC product engineered to express human interferon alpha-2 under the control of the tyrosine kinase with Ig and EGF homology domains 2 (TIE2) promoter via lentiviral gene transfer. The aim is to activate anti-tumor immunity by having TIE2-positive macrophages present in the tumor microenvironment (TME) express interferon. Clinical trials for glioblastoma have reported increased infiltration of TIE2-positive macrophages into tumors, altered gene expression in the TME, and prolonged survival compared to historical controls. Glioblastoma remains a disease with an extremely poor prognosis and is difficult to treat, one reason being the difficulty in delivering drugs to the tumor. It is hoped that the HSC-based approach, enabling the efficient delivery of therapeutic proteins, will improve treatment for this devastating disease.49–51
Potentially, therapeutic proteins may also include antibodies. Luo et al. demonstrated that using a lentiviral vector to introduce a gene encoding an HIV-1 gp120 neutralizing antibody into human HSPCs resulted in induced B cells and plasma cells expressing the antibody protein. 52 This study suggests the technological potential for sustained in vivo production of therapeutic antibodies through a mechanism distinct from vaccines. While antibody therapeutics generally require repeated administration to maintain efficacy, cell therapeutics—which possess entirely different pharmacokinetic profiles from protein therapeutics—hold the potential to significantly reduce the number of required doses. HSC-based therapeutics, in particular, have demonstrated the potential for lifelong therapeutic effects due to their persistence. Luna et al. experimentally demonstrated this potential using clinically established antibodies: an anti-proprotein convertase subtilisin kexin 9 antibody for treating hypercholesterolemia and an anti-tumor necrosis factor antibody for treating autoimmune diseases. They introduced genes encoding these antibodies into human HSCs and transplanted them into immunodeficient mice, confirming that the HSCs engrafted normally. Since the reconstruction of the human immune system in immunodeficient mice is incomplete, they used a system where murine HSCs with introduced antibody genes were transplanted into immunocompetent mice. This confirmed that these therapeutic antibodies were produced at levels sufficient to exert therapeutic effects in vivo. For safety considerations, a system capable of appropriately adjusting antibody levels appears necessary. 53 This approach could provide a novel delivery modality for therapeutic antibodies targeting chronic diseases.
“Molecular shielding” is an emerging concept: it means genetically modifying cells (via deletion, mutation or epitope engineering on target molecule) to confer resistance to molecularly targeted drugs, including chimeric antigen receptors (CARs).54–56 This can be a highly useful concept in cell therapy, particularly compelling for HSCs because shielding the stem cell protects all its progeny. A pioneering example of this technology is Tremtelectogene empogeditemcel (Trem-cel), an allogeneic HSC that uses the CRISPR/Cas9 system to delete the CD33 gene. 57 CD33 is a well-established target molecule for acute myeloid leukemia (AML), widely expressed in malignant myeloid cells. However, it is also expressed in normal HSPCs. Consequently, CD33-targeted therapies result in on-target toxicity, including cytopenia and myelosuppression, presenting a narrow therapeutic window between effective and toxic doses. Importantly, it has been demonstrated that CD33 deletion does not affect hematopoietic stem cell function and allows for the reconstitution of a functionally normal hematopoietic system.54,58 This reconstituted hematopoietic system lacks CD33 and therefore exhibits resistance to CD33-targeted therapies. A clinical trial combining Trem-cel with gemtuzumab ozogamicin (GO), an anti-CD33 antibody-drug conjugate, for patients with AML was conducted by VOR Bio. 59 In this trial, Trem-cel was transplanted first, followed by GO administration after engraftment. Preliminary data published to date indicate normal engraftment and increased GO exposure in the Trem-cel group. Furthermore, the neutropenia and thrombocytopenia frequently observed with GO administration were not seen, suggesting that CD33 deletion can enhance therapeutic efficacy of GO.60,61 Since VOR announced a significant strategic shift recently, and Trem-cel has now disappeared from its pipeline, it is unclear whether Trem-cel will continue to be pursued in the future. 62 However, despite the limited number of cases, the clinical confirmation that CD33-negative human HSCs engraft and confer resistance to GO holds significant implications for considering the application of molecular shielding through HSC engineering in humans.
While CD33 deficiency is thought not to affect HSC function, this approach is limited for genes whose deficiency impairs HSC or immune cell function. For target molecules where simple deletion is not feasible, more sophisticated techniques have been developed. CD45 is a pan-leukocyte marker molecule expressed on nearly all blood cells except erythrocytes and platelets. It is known that CD45 deficiency causes abnormalities in T cell development and immunodeficiency.63–65 CD45 is expressed in virtually all blood cancers except multiple myeloma (MM).66,67 While the extensive expression pattern of CD45 in the normal hematopoietic system posed a hurdle to targeting it therapeutically, Wellhausen et al. and Garaudé et al. independently demonstrated that editing the epitope of CD45 antibodies in HSCs shields the HSCs and progenies, and enables antibody-drug conjugates (ADCs) and CAR-T cells to exert cytotoxic activity selectively against cancer cells. They independently identified epitopes on their respective anti-CD45 monoclonal antibodies (BC8 and MIRG451) and pinpointed mutations that inhibit antibody binding without affecting CD45 function. Furthermore, using base editing technology to introduce mutations at those sites into human HSCs resulted in the emergence of an HSC population unrecognized by the antibodies. They evaluated different antibodies and CD45 mutations, demonstrating in each case that the introduced mutations did not affect HSC function.68,69 The normal hematopoietic system reconstituted by the modified HSCs exhibits resistance to CD45-targeted therapy, enabling selective treatment of cancer cells. The lack of target molecules that accurately distinguish normal cells from cancer cells is a fundamental problem in blood cancer therapy. However, the molecular shielding approach can generate cancer-cell-selective therapeutic targets. This strategy could provide a new therapeutic option for treating intractable hematological malignancies.
In Vivo HSC Genetic Modification
Another compelling idea involves generating therapeutic cells in vivo from genetically modified HSCs. Introducing genes encoding CARs into HSCs and allowing them to differentiate in the body enables the sustained production of CAR-immune cells. This approach could address one of the key limitations of current CAR-cell therapies: the challenge of in vivo persistence. Furthermore, while current cell therapies use only a single type of immune cell, modifying HSCs could enable the simultaneous production of multiple therapeutic immune cell types within the body, potentially leading to more effective cell therapies.
Studies applying this concept to anti-HIV therapy have recently been published, using a CAR construct containing the extracellular domain of CD4 to interact with the HIV envelope. The CD4-CAR gene was transduced into HSCs to differentiate CD4-CAR-immune cells in vivo. Interestingly, HSC-derived CD4-CAR cells demonstrated superior CAR-T cell proliferation and viral control capabilities compared to conventional peripheral blood-derived CAR-T cells in a humanized mouse model.60,61 Furthermore, studies using non-human primates showed that HSC-derived CAR-T cells exhibit superior tissue infiltration compared to peripheral blood-derived CAR-T cells. In a separate paper by the same group, experiments using non-human primates demonstrated that CAR-positive cells from multiple lineages—including T cells, B cells, and myeloid cells—can differentiate from modified stem cells. This is achieved by using a universal promoter, which is expected to enhance the immune response against viral antigens compared to approaches utilizing only T cells. The authors suggest that this approach is applicable not only to viral infections but also to cancer, where CAR cell therapy is currently mainly and actively applied.70–72
Recently, Louvet et al. reported that by expressing anti-B-cell maturation antigen (BCMA) CAR—a well-validated target molecule in MM—under natural killer (NK) cell-selective promoter control, HSCs can be utilized as “CAR-NK factories” enabling continuously producing anti-BCMA-CAR-NK cells in vivo. The authors demonstrated that in their system, CAR is expressed selectively on NK cells and not on other cell types. Furthermore, using a xenograft mouse model with MM cells, they showed anti-tumor activity. MM is known as a disease that is difficult to cure despite the existence of many treatment options. However, this study suggests the potential to achieve a functional cure using a highly sustainable method utilizing stem cells. 73
A similar idea can be applied to autoimmune diseases. Ng et al. generated T-cell receptor (TCR) retrogenic mice by introducing specific TCRs derived from T cell clones obtained from amino acid (YFAK) copolymer-treated mice into murine HSPCs via retroviral transduction and subsequent transplantation. 74 They found that T cells differentiating in these mice produced high levels of IL-10, and the mice exhibited resistance to experimental autoimmune encephalomyelitis induction. 74 Orchard Therapeutics developed an ex vivo therapeutic approach for autoimmune diseases by differentiating CAR-Tregs that express self-antigen-selective CARs from genetically modified HSCs. 75 These efforts could lead to a novel therapeutic approach for severe chronic autoimmune diseases such as multiple sclerosis.
Gene Expression Control Technologies in HSC Gene Therapy
In cell and gene therapy, controlling the expression of therapeutic genes is critically important. Inadequate or excessive expression of a therapeutic gene can reduce efficacy and/or cause safety issues. Therefore, for each product, the gene expression control system is designed to provide the desired gene expression intensity and selectivity. 76
Some gene therapies employ constitutive gene expression systems designed to provide robust expression across a wide range of cell types. Early gene therapies primarily utilized the promoter activity of LTR elements of retroviral origin. However, viral-derived sequences may be prone to silencing and influence endogenous gene expression around the insertion site. It is now more common to generate a gene expression cassette under the control of an appropriate promoter in the context of an SIN vector. Promoters providing constitutive expression include those from mammalian housekeeping genes such as phosphoglycerate kinase and elongation factor 1-alpha (EF-1α). 77 Given that current vector systems involve random insertion into the genome, using mammalian-derived promoters and short-range enhancer sequences is generally considered safer. 78
When considering HSC therapy, where therapeutic cells are produced through differentiation of transplanted autologous HSCs, inappropriate transgene expression can affect the stem cell properties of HSCs or the development of effector cells, potentially leading to unexpected safety risks. Therefore, compared to conventional cell therapies based on a single cell type, highly sophisticated and precise gene expression control is required.
In the aforementioned TemferonTM, TIE2 was identified as a gene selectively expressed in myeloid cells within TME of glioblastoma. The therapeutic gene interferon alpha 2 (IFNa2) was linked downstream to the TIE2 promoter. Furthermore, to prevent transgene expression in HSPCs, a target sequence for microRNA126, which is highly expressed in HSPCs, was incorporated. 50
Colamartino et al. designed a synthetic promoter selective for T cells to generate CAR-T cells in vivo from genetically modified CD34-positive cells. They confirmed the cell type specificity by transducing the green fluorescent protein (GFP) gene under the control of the promoter into human peripheral blood mononuclear cells to see T cell-selective fluorescent protein expression. 79 Such cell-selective promoters would be critical components for achieving therapeutic cell production from genetically modified HSCs. While fluorescent proteins are commonly used as reporter genes to visualize transcriptional control in promoter studies, gene expression is significantly influenced by other post-transcriptional, translational, and post-translational cell factors. Therefore, verification and optimization to ensure sufficient expression levels and selectivity using the actual therapeutic gene is of crucial importance.
Gene modification technology for HSCs using lentiviral vectors has achieved efficiency and safety levels sufficient for regulatory approval, offering patients a favorable risk-benefit ratio for severe genetic disorders. However, the risk of unexpected genotoxicity due to uncontrolled transgene insertion cannot be completely eliminated. Also, from the perspective of transgene expression control, utilizing endogenous gene expression regulatory mechanisms appears effective. For these reasons, genome-site-specific insertion of therapeutic genes may become the mainstream approach in the future. Welty et al. reported that knocking the CAR gene into the CD33 locus enables the stable, long-term production of CAR macrophages (CAR-M) from modified HSCs. Fully differentiated myeloid cells generally have a short lifespan, which is one challenge when utilizing myeloid cells for therapeutic purposes. Production from HSCs may overcome this limitation. In this report, CAR-M targeting human epidermal growth factor receptor 2 (HER2), produced from modified HSCs, was shown to infiltrate tumors and significantly increase T-cell infiltration. 80 Nonviral knock-in into the CD33 locus could be an effective approach when stable transgene expression is required in myeloid-lineage cells.
Safety Switches
Genetically modified HSCs maintain their effect by persisting long-term in the patient and committing to hematopoiesis. Reports of long-term follow-up after lentiviral HSC gene therapy indicate sustained efficacy for over 10 years. Potentially lifelong persistence of effect is one of the most important characteristics of HSC-based gene therapies, but it simultaneously raises safety concerns in case the therapeutic cells or genes show unwanted behavior. Technologies that control cell survival via external cues have the potential to mitigate these concerns. Various mechanisms for eliminating cells (suicide switches) have been devised, and their use could enable safer applications. A well-known technology is based on the herpes simplex virus thymidine kinase (HSV-TK). HSV-TK induces cell death by phosphorylating and converting the normally inactive substrate ganciclovir (GC) into a cytotoxic nucleotide analogue.81,82 However, the HSV-TK/GC system showed several limitations, including slow and incomplete cell death depending on the cell type and immunogenicity due to its viral origin. 81 Therefore, a system utilizing caspase, a mammalian endogenous apoptosis-inducing factor, was devised. The most widely used system is iCaspase9/Ap1903.83,84 This consists of a fusion protein of a drug-binding domain of FK506 binding protein (FKBP) and a catalytic domain of human caspase9; the small molecule AP1903 dimerizes FKBP, thereby inducing apoptosis in cells. iCaspase9 is based on human-derived caspase-9, offering the advantages of low immunogenicity risk, rapid apoptosis induction, and minimal dependence on the cell’s proliferative state. Additionally, similar technologies utilizing caspase-9 have been reported, such as a rapamycin-activated caspase-9 (RapaCasp9) system utilizing rapamycin as a dimerizing agent. 85 Although challenges remain, such as the increasing complexity of cell engineering and the need to verify switch functionality in clinical trials, suicide-switching technology is expected to enhance the safety of HSC-based therapies and promote their clinical application.
Technical Improvements for Expanding Applications
To date, applications of HSC gene therapy have been limited to severe congenital diseases. Technological developments in peripheral fields have the potential to significantly expand the scope of application for this technology. (See Fig. 2)
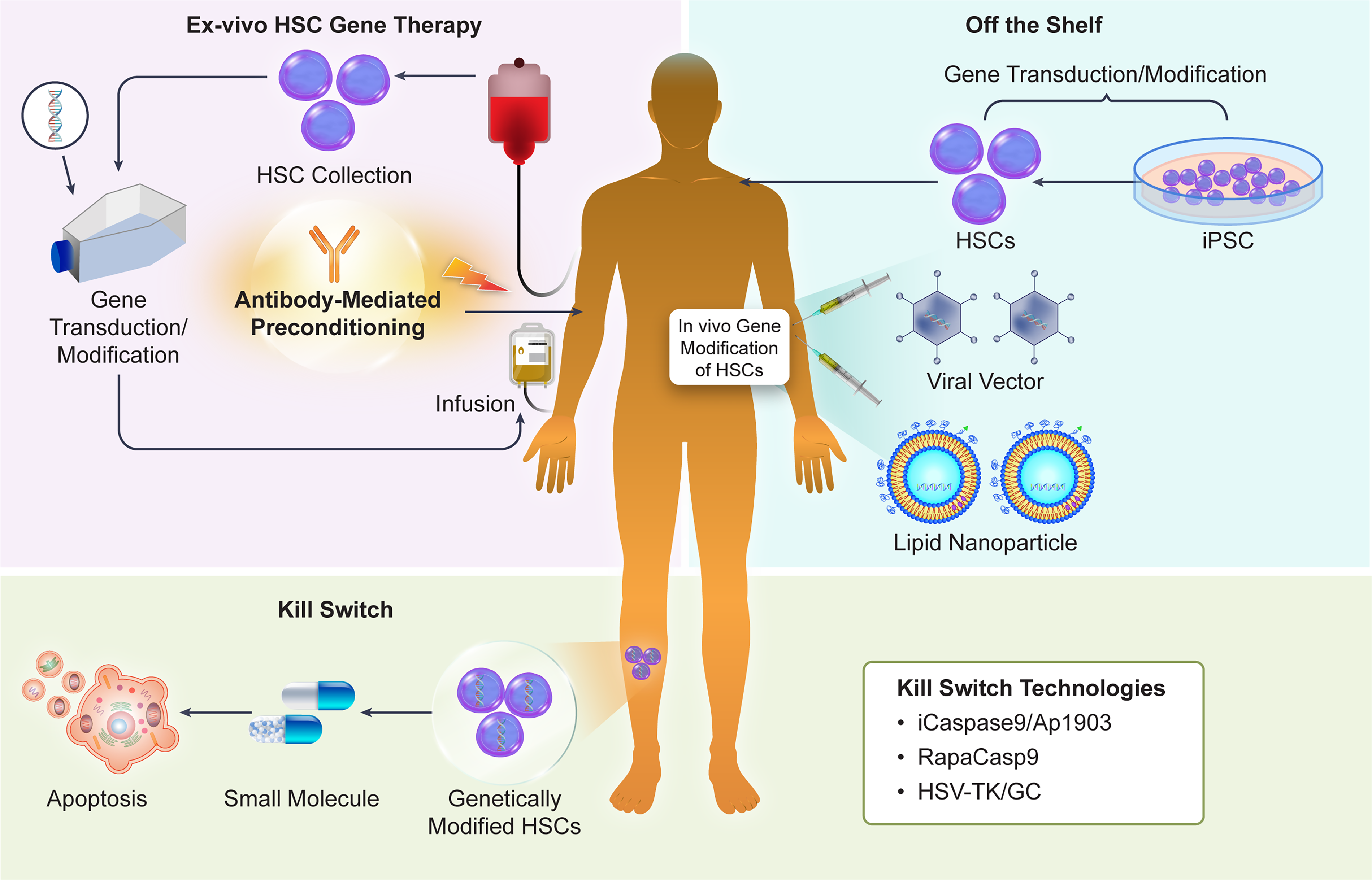
Technologies expanding the application of HSC-based cell and gene therapies.
Pre-conditioning for HSCT
For exogenously infused HSCs to engraft in the recipient’s bone marrow and initiate hematopoiesis, even in autologous HSCT, preconditioning with myeloablative chemotherapy or systemic irradiation is required. Conditioning removes the recipient’s HSCs and opens the HSC microenvironment, known as the stem cell niche, to allow the engraftment of transplanted cells. However, systemic toxicity from conditioning agents and infections due to compromised immune function are frequent complications. 86 HSCT, including managing complications, is established as a standard procedure for a number of life-threatening disorders, including hematological malignancies and certain autoimmune diseases. However, when applying HSCT to less severe diseases and/or non-malignant conditions, the toxicity of conditioning may pose a significant hurdle. Also, for HSC gene therapy, pre-transplant conditioning offers a favorable risk-benefit balance in the case of severe diseases but not necessarily in the treatment of less severe ones. An optimized conditioning regimen that minimizes acute toxicity and genotoxicity while preserving sufficient myeloablation is being explored. 87 In particular, “cleaner” conditioning using monoclonal antibodies has been proposed to replace busulfan or total body irradiation. Antibodies targeting surface antigens expressed on HSCs, such as KIT proto-oncogene, receptor tyrosine kinase (c-KIT, CD117) and myeloproliferative leukemia protein (c-MPL), have been shown to clear the niche without the need of chemotherapy, allowing engraftment of exogenous HSCs.88–90 In particular, patients with Fanconi anemia (FA) exhibit heightened sensitivity to DNA damage and carry increased risks of tissue injury and secondary malignancies, making the use of less genotoxic conditioning a necessary prerequisite. A protocol without radiation or busulfan for patients with FA using the anti-CD117 antibody briquilimab is being tested clinically, with promising results. 91 Such protocols could prove useful for applying HSC gene therapy to broader indications.
Off-the-shelf approaches
Current HSC gene therapies, which process the patient’s own cells, share the high costs and logistical challenges associated with “N = 1” manufacturing seen in other autologous cell products like CAR-T. This could be a barrier to the widespread application of this technology. Developing off-the-shelf products that can be pre-manufactured and stockpiled is a potential alternative for achieving broader adoption of gene therapy technology.
Induced pluripotent stem cells (iPSCs) have the potential to serve as a source for an unlimited supply of HSCs that are compatible with the patient and independent of donors. 92 Protocols for inducing HSCs from iPSCs are basically designed to mimic the developmental process in vivo. Scapin et al. reported that using PIEZO1 signaling, which mimics the mechanical cues promoting HSC development from hematopoietic endothelial cells in vivo, they could induce long-term HSCs (LT-HSCs) from human iPSCs that engraft long-term in mice and reconstitute the hematopoietic system without exogenous gene transfer. 93 As the HLA mismatch between donor and recipient increases the risk of transplant rejection and GvHD, to make off-the-shelf iPSC-derived HSCs a viable therapeutic option, it would be necessary to construct an iPSC bank covering HLA types compatible with many recipients. In multiple countries, efforts are underway to generate clinical-grade iPSC banks covering the HLA types of each national population.94–98 These efforts could form the basis for off-the-shelf HSC-based therapies.
In vivo genetic manipulation of HSCs
Another important strategy aimed at broadly implementing HSC gene therapy involves direct gene delivery to, or gene editing of, HSCs in vivo. This approach involves administering gene delivery vectors, such as viral vectors or lipid nanoparticles (LNPs), directly to the patient to introduce genes and/or perform gene editing in HSCs within the bone marrow. By directly modifying the patient’s own HSCs, it may be possible to avoid the genotoxic pre-conditioning described above. Breda et al. demonstrated that CD117 antibody-targeted LNPs can deliver mRNA to LT-HSCs in vivo. Furthermore, they proved in vitro that their LNP can transport Cas9 adenine base editing enzymes and guide RNA, correcting the pathogenic mutation of sickle-cell disease to a non-pathogenic form. 99 Lieber’s group demonstrated that intravenous administration of a helper-dependent Ad5/35 vector enables in vivo gene delivery and genome editing of HSCs.100–102 HD-Ad5/35++ is thought to target quiescent HSCs via the CD46 molecule. It is also characterized by its large cargo size of 35 kb. A key feature of this methodology is the in vivo selection of genetically modified HSCs using low-dose cytotoxic drugs and a drug resistance gene carried by the vector. The authors demonstrated that this approach enables the expansion of genetically modified HSCs in non-human primates with acceptable toxicity. 102 Ensoma is currently advancing the development of in vivo HSC gene therapy based on this technology. 103 Other companies are pursuing non-viral gene delivery to HSCs, at different stages of pre-clinical or clinical development. In vivo HSC gene editing is rapidly advancing and undoubtedly one of the most important topics in this area (reviewed in Ball et al. 2025), 104 to the point that it is difficult to cover comprehensively in this article. In the future, in vivo gene delivery and gene editing are expected to be applied to a wide range of diseases and make HSC gene therapy less complex, less expensive, and more beneficial to patients.
Conclusions
HSC gene therapy has seen rapid progress over the past few decades. Despite a mixed history of successes and failures during its early days, incremental technical improvements, from the development of safer vectors to tailored gene expression and robust manufacturing technology, have culminated in efficacious treatments for a number of devastating monogenic diseases, offering the hope of a one-time definitive cure. However, considering the unique nature of HSCs, which supply all types of immune cells, its applications are not limited to congenital disorders. HSC gene therapy holds the potential to provide treatments for diseases with more complex pathogenesis, such as cancers and autoimmune disorders, which cannot be effectively treated with conventional modalities. Oncology is often the field where new therapeutic modalities are first applied. In particular, hematological malignancies, where HSCT is already integrated into standard treatment, are highly likely to be the first area where genetically engineered HSC therapeutics will be applied. Continuous supply of therapeutic molecules/cells and conferring resistance to toxic agents through genetic modification (shielding) are great examples of concepts only possible with genetically engineered HSCs. The development of new therapies utilizing these concepts is anticipated. As discussed in this short review article, considering the rapid technological innovations underway to mitigate or resolve risks specific to HSCs—such as more precise genome editing, gene expression control, and conditioning regimens—it is anticipated that the scope of HSC-based cell and gene therapies applications will greatly expand in the near future and increasingly utilized to save or improve the life patients suffering from intractable diseases.
Footnotes
Acknowledgment
The authors thank their colleagues for their critical feedback.
Author Disclosure Statement
M.K. is an employee of Kyowa Kirin Co., Ltd. F.M. is an employee of Orchard Therapeutics.
Funding Information
No competing financial interests exist.