
Abstract
Introduction
Maraviroc is the first orally bioavailable, noncompetitive CCR5 chemokine coreceptor antagonist approved for the management of chronic human immunodeficiency virus (HIV) infection. 1 By binding to CCR5 receptors on the host cell membrane, maraviroc triggers conformational changes in the ECL2 region of CCR5 that prevents interaction of the coreceptor with the V3 crown 1 –3 of gp120. As a result, maraviroc demonstrates 2,3 selective and potent in vitro activity against CCR5-tropic HIV isolates, with a 90% inhibitory concentration of 2.0 nmol/L. Given its in vitro potency, novel mechanism of action and the consequent lack of cross-resistance with established classes of antiretroviral (ARV) agents, maraviroc has emerged as a promising candidate for the management of treatment-experienced patients with limited options. Clinical corroboration of the in vitro antiretroviral potency of maraviroc was subsequently provided by randomized trials performed in treatment-experienced patients. Specifically, significantly greater reductions in plasma viral load were observed in ARV-experienced patients randomized to receive maraviroc with optimized background therapy (OBT) relative to patients randomized to receiving placebo and OBT in both the MOTIVATE 1 and 2 studies. 4 –6 A notable ancillary finding of the MOTIVATE studies was a robust immunologic recovery associated with maraviroc that was independent of its effect on plasma viral load. 7 Postulated mechanisms for the immunologic enhancing effect of maraviroc include inhibition of the trafficking of CCR5+ T-lymphocytes between the circulation and peripheral tissues and reduced immune activation resulting from the antagonism of the CCR5 receptor. 8 Interest in exploiting this property of maraviroc has understandably emerged, particularly for the management of ARV-experienced patients in whom immunologic recovery may be blunted despite full virologic suppression.
However, an important caveat in the interpretation of findings from the MOTIVATE trials is that the OBT available for use in these studies does not mirror current clinical practice. Most notably, raltegravir, a commonly deployed agent when treating patients with drug-resistant HIV, was not permitted in the MOTIVATE trials, given its investigational status at that time. Because pharmacokinetic studies have confirmed that maraviroc does not have deleterious effects on raltegravir exposure and the 2 agents are not expected to exhibit antagonistic pharmacodynamic effects when used as components of a regimen for treatment-experienced patients, combined use of these agents is appealing for managing this subgroup of HIV-infected patients. 9 However, it is unclear whether the addition of maraviroc to patients receiving contemporary OBT which includes raltegravir confers additional immunologic benefits to patients when compared with raltegravir-based therapy alone. The purpose of our study was to compare the immunologic effectiveness of raltegravir–maraviroc (R+M+)-based regimens with raltegravir-based regimens that do not include maraviroc (R+M−) in treatment-experienced patients in clinical practice.
Methods
We conducted a retrospective cohort study of HIV-infected adults who were receiving either R+M+- or R+M−-based antiretroviral therapy (ART) at an inner city primary care clinic in Toronto, Ontario, Canada. Patients were eligible for inclusion in the analysis if they were ARV experienced with a history of treatment failure, had a baseline CD4 count within 6 months prior to initiating either R+M+- or R+M−-based therapy, and had at least 1 CD4 count measurement performed following the initiation of therapy with either R+M+ or R+M−. Antiretroviral agents used in conjunction with R+M+ or R+M− were selected based on treatment history and genotypic resistance testing at the discretion of the treating physician. Ethics approval was obtained from the local Institutional Review Board prior to analysis.
Statistical Analysis
Demographic characteristics of patients receiving R+M+ or R+M− were described using frequencies and proportions for categorical variables and medians and interquartile ranges for continuous variables. Baseline characteristics of the 2 groups were compared using the Wilcoxon rank sum test for continuous variables and the chi-square test or Fisher Exact test for categorical variables.
The primary end point of the study was the rate and degree of change in CD4 count within 24 months of initiation of R+M+- or R+M−-based therapy. The baseline CD4 count was the most recent CD4 count prior to the initiation of R+M+ or R+M− therapy but not more than 6 months before the initiation of therapy. For patients with fewer than 24 months of follow-up data available, the end of follow-up was defined as either the date of termination of maraviroc or raltegravir for patients receiving R+M+ or the date of termination of raltegravir for patients receiving R+M−. Longitudinal CD4 counts were analyzed using a linear mixed model with a spatial correlation structure to accommodate the correlation of CD4 measurements within patients. Covariates included the main effects of treatment, time (months of follow-up), and the interaction term of treatment and time, with additional terms for baseline age, years of being HIV positive, baseline HIV viral load, use of a nonnucleoside reverse transcriptase inhibitor (NNRTI), and use of a ritonavir (RTV)-boosted protease inhibitor (PI). Of primary interest was the effect of the interaction between treatment and months of follow-up which measures the difference in patterns of changes in the mean CD4 over time between the 2 treatment groups.
For the subset of patients who had detectable baseline viral load, the log-rank test was used to compare time to virologic suppression between the 2 treatment groups.
The primary toxicity end points of interest were the occurrence of grade 3 to 4 elevations in serum alanine transaminase (ALT), creatine kinase (CK), cholesterol, or triglycerides during the first 24 months of treatment, according to the National Institute of Allergy and Infectious Diseases Division of AIDS toxicity grading tables. 10
All statistical analyses were performed using SAS Version 9.2 (SAS Institute, Cary, North Carolina).
Results
Patient Characteristics
A total of 182 patients were identified as having taken either R+M+- or R+M−-based therapy. Twenty-six patients were excluded from the study because of missing CD4 count either at baseline or during follow-up. One hundred and fifty-six patients were included in the analysis, 32 on R+M+ and 124 on R+M−. The median number of days of follow-up and the average number of CD4 cell measurements were 713 days (interquartile range [IQR] 453.5-731) and 9 (IQR 4-11.5) for R+M+ and 730 days (IQR 429.5-731) and 8 (IQR 5-11) for R+M−. Among patients on R+M+, 16 had ≥24 months of follow-up, 8 had <24 months of follow-up and are still on treatment, and 8 stopped treatment before 24 months of follow-up. Among patients on R+M−, 71 had ≥24 months of follow-up, 33 had <24 months of follow-up and are still on treatment, and 20 stopped treatment before 24 months of follow-up.
The baseline characteristics of the 2 groups are summarized in Table 1. Almost all participants were male, with a median age of 50 (IQR 45.3-55.3) and a median of 16 years (IQR 9.5-19.3) since their first HIV-positive test. The mean CD4 count at baseline was higher among patients on R+M+ than those on R+M− but the difference was not statistically significant (463.8 vs 442.3 cells/mm3; P = .67). The 2 groups differed with respect to the number of ARV medications in regimen (5.5 [IQR 5-6.5] for R+M+ vs 4 [IQR 3-5] for R+M−; P < .001). No differences were observed in the nature of the OBT antiretroviral drugs used between the 2 groups. The proportion of patients receiving concomitant enfuvirtide was similar between the 2 groups.
Summary of Baseline and Demographic Characteristics in the Study of Raltegravir and Maraviroc
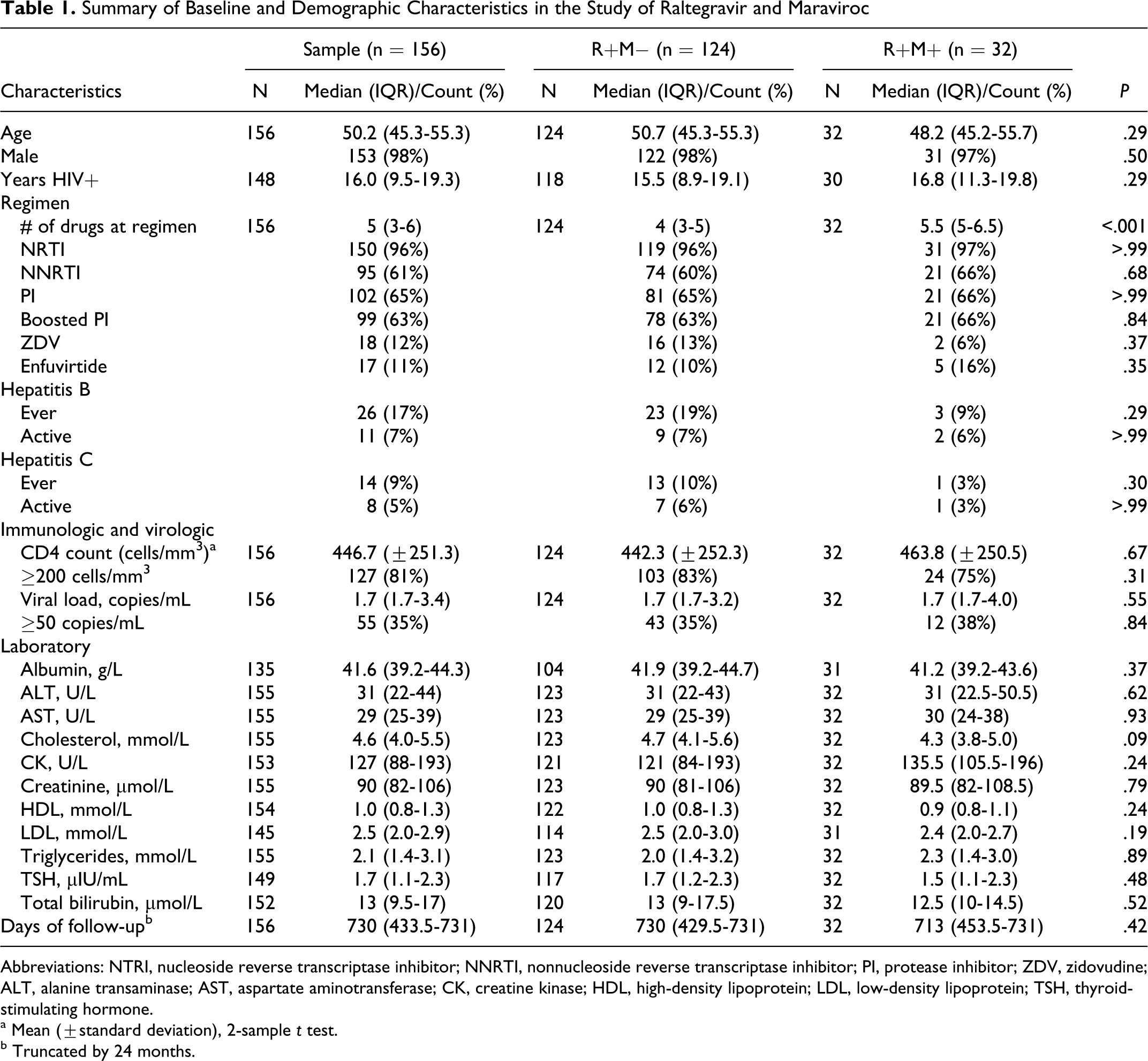
Abbreviations: NTRI, nucleoside reverse transcriptase inhibitor; NNRTI, nonnucleoside reverse transcriptase inhibitor; PI, protease inhibitor; ZDV, zidovudine; ALT, alanine transaminase; AST, aspartate aminotransferase; CK, creatine kinase; HDL, high-density lipoprotein; LDL, low-density lipoprotein; TSH, thyroid-stimulating hormone.
a Mean (±standard deviation), 2-sample t test.
b Truncated by 24 months.
Immunologic Effectiveness
The mean CD4 count by treatment group and month of follow-up is shown in Figure 1. In univariate mixed models, no significant difference was observed between patients treated with R+M+- or R+M−-based therapy in rates of change in the CD4 count over time (−0.08 cells/mm3, P = .98; Table 2). Variables associated with a decrease in CD4 during follow-up in univariate models included the baseline viral load (P < .0001) and the number of ARV medications in the regimen (P < .0001; Table 2). In multivariable mixed models, only baseline viral load remained significantly associated with CD4 change during follow-up. No difference between R+M+ and R+M− was observed (P = .81; Table 3).

Mean CD4 count by month of follow-up
Univariate Mixed Models on Correlated CD4 Repeated Measurements

Abbreviations: NTRI, nucleoside reverse transcriptase inhibitor; NNRTI, nonnucleoside reverse transcriptase inhibitor; PI, protease inhibitor; ZDV, zidovudine; VL, viral load; ALT, alanine transaminase.
Multivariable Mixed Models for Longitudinal Change in CD4 Count over Follow-Up

Abbreviations: NTRI, nucleoside reverse transcriptase inhibitor; NNRTI, nonnucleoside reverse transcriptase inhibitor; PI, protease inhibitor.
Among the 55 patients with detectable viremia at baseline, there was no significant difference in the median time to virologic suppression between treatment patients receiving either R+M+ or R+M− (84 vs 79 days, P = .26; Figure 2).

The Kaplan-Meier plot for time to VL suppression in the subset of patients who had detectable baseline VL. VL indicates viral load
Toxicities
The proportions of patients who experienced at least 1 grade 3 or 4 elevation in various laboratory parameters were not significantly different between treatment groups (Table 4).
Number and Percentage of Patients Who Had Ever Experienced at Least 1 Grade 3 to 4 Toxicity during the Study Period

Abbreviations: ALT, alanine transaminase; AST, aspartate aminotransferase; CK, creatine kinase; LDL, low-density lipoprotein.
Discussion
Our findings demonstrate that CD4 count recovery was similar among patients receiving either R+M+- or R+M−-based therapy, suggesting that the use of maraviroc in patients receiving OBT that includes raltegravir does not confer additional immunologic advantages during a 24-month period of follow-up. Our findings corroborate those of a smaller study, in which the median increases in CD4 count after 6 months of treatment among patients receiving raltegravir with (n = 11) and without (n = 43) maraviroc were 84 cells/mm3 (range −117 to +307) and 68 cells/mm3 (range −405 to 343), respectively. 9
The impact of maraviroc on immunologic recovery in treatment-experienced patients has been evaluated in several trials and small reports, with conflicting results. Our findings add to the existing body of knowledge regarding the optimal use of maraviroc for treatment-experienced patients from an immunologic perspective. In the MOTIVATE studies, a significant immunologic advantage was afforded by maraviroc when compared with placebo, in that the median increases in CD4 count among patients randomized to once daily (QD) maraviroc, twice daily (BID) maraviroc, or placebo were 92, 103, and 24 cells/mm3, respectively (P < .05). 7 The difference in CD4 gain remained significant following adjustment for predictors of immunologic recovery and when the subset of patients attaining an undetectable viral load was examined. Importantly, the heightened increase in CD4 count was associated with a significantly longer time to new AIDS-defining events among patients receiving maraviroc. These findings were supported by 24-week results from the A4001029 study of treatment-experienced HIV-positive patients with viruses of dual/mixed, CXCR4, or undetermined tropism, in whom greater gains in CD4 counts were observed among patients receiving maraviroc relative to placebo (62 vs 36 cells; P = .04). 11 Finally, a meta-analysis of clinical trial data demonstrated that, relative to nonuse, use of CCR5 antagonists was associated with an added CD4 count increase of approximately 30 cells/mm3 after controlling for virologic suppression. 12 Taken together, these data suggest that CCR5 antagonists play a critical role in the management of treatment-experienced patients, particularly the subgroup of patients who attain incomplete immunologic recovery despite virologic suppression. However, as previously mentioned, several limitations of the aforementioned trials have limited the generalizability of the inferences to current clinical practice. Notably, the availability of darunavir, etravirine, and raltegravir has amplified the potency of regimens available for patients with extensive underlying ARV resistance and otherwise few treatment options. The OBT assembled for patients in the MOTIVATE studies is therefore likely to have been virologically and immunologically inferior to what is presently constructible for a similar cohort of patients. In addition, the 24-week advantage conferred by maraviroc in the A4001029 study had dissipated by week 48, in that there was no significant difference in CD4 count response between maraviroc and placebo groups at this point in follow-up. 11 In addition, recently published data are casting additional doubt on the benefits of intensifying regimens with maraviroc for patients who are immunologic nonresponders. Findings from a second meta-analysis suggested that maraviroc was not associated with more robust immunologic recovery in treatment-experienced patients relative to other newer ARV medications, such as raltegravir and darunavir. 13 Furthermore, in a small report of 9 patients, the addition of maraviroc to regimens associated with sustained virologic suppression did not yield a significant change in CD4 count after 25 weeks of therapy. 14 Similarly, in a single-arm pilot study of 34 patients with undetectable viral load and a median baseline CD4 count of 153 cells/mm3, the addition of maraviroc to the existing virologically suppressive regimen for 24 weeks was not associated with a prespecified clinically significant increase in CD4 count of at least 20 cells/mm3, despite concomitant reductions in CD4 and CD8 cell activation. 15 However, these findings are obviously limited by the small sample sizes of the reports and the observational nature of the designs.
The results of our study contribute to the emerging body of knowledge regarding the optimal use of maraviroc for the management of HIV-infected individuals. While maraviroc remains an important option for the management of treatment-experienced patients with CCR5-tropic virus, particularly in the setting of extensive resistance to PIs and/or NNRTIs, our analyses do not support a role of maraviroc as an immunologic enhancing agent among patients who are receiving raltegravir-based therapy. However, several limitations of our work merit emphasis when interpreting our findings. Notably, there was considerable heterogeneity among patients receiving maraviroc in terms of viral tropism, and these data were unknown for the 35% of R+M+ patients who had an undetectable viral load at the time of maraviroc initiation. Therefore, our findings may not be reproducible in a larger study of patients harboring CCR5-tropic virus in whom the benefits of maraviroc are expected to be maximized. In addition, over 80% of patients in our study had baseline CD4 counts above 200 cells/mm3. Therefore, we cannot extrapolate our findings to patients with more advanced degrees of immune suppression prior to initiating treatment. Finally, our sample size was small, and our study was retrospective and nonrandomized in nature. Although we adjusted our analyses for important confounders, we cannot rule out the introduction of bias through unmeasured confounding variables. Despite these limitations, however, our study has numerous strengths, including a relatively large sample of patients receiving maraviroc, a repeated measures analysis, and a focused, clinically relevant question directly applicable to current HIV practice.
In conclusion, our study provides important comparative data of the immunologic effectiveness of R+M+- and R+M−-based regimens in a clinical setting. Following multivariable adjustment, no significant difference was observed between the 2 treatment arms in terms of the rate of change in CD4 count over 24 months of follow-up. Although our patients were not randomized to individual treatments, this study represents the only available analysis evaluating the immunologic contribution of maraviroc to regimens which include raltegravir. Additional data from observational studies or controlled trials in patients with more advanced immunosuppression and/or harboring CCR5-tropic virus exclusively would further clarify the role of maraviroc in the management of treatment-experienced HIV-infected patients receiving concurrent treatment with raltegravir.
Footnotes
Acknowledgment
The authors thank the patients and staff at the participating site for their contribution to this work.
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: TA has received unrestricted research grants from GlaxoSmithKline, Pfizer and Merck Inc. for unrelated research projects. CK has been on advisory boards and/or received unrestricted research funds from Merck, Bristol-Myers-Squibb, Pfizer, Viiv, Abbott and Gilead. JB has received honoraria for consulting work from Abbott, ViiV, Tibotec, Merck, Eli Lilly, and Gilead. ML has received unrestricted research grants for other projects from, and has acted as a speaker and advisor for, Abbott Canada, Merck Frosst, Pfizer, Bristol-Myers Squibb, Tibotec, Boehringer Ingelheim, and GlaxoSmithKline. FC has acted as a speaker and advisor for Abbott, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead, GlaxoSmithKline, Lilly, Merck, Pfizer, Solvay, Tibotec and ViiV. GS has received speaker fees and advisory board fees from Merck, Abbott, Viiv and Tibotec/Jansen. DF has received honoraria for consulting work from Abbott, ViiV, Tibotec, Merck, Bristol Myers Squibb, and Gilead.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: TA is supported by a postdoctoral fellowship award from the Ontario HIV Treatment Network. JMR received Career Scientist support from the Ontario HIV Treatment Network. MRL is supported by a New Investigator Award from the Canadian Institutes of Health Research.