
Abstract
The etiology and pathogenic mechanisms underlying Sjögren’s syndrome (SS) remain unclear. Recent studies have emphasized that the specific autoantibodies that occur in a high proportion of patients with SS may provide important insights into the circumstances that initiate and propagate tissue damage in this disease. Although autoantigens targeted in systemic autoimmune diseases share little in common in terms of structure, subcellular distribution, or function in normal cells, these molecules are unified by becoming clustered and concentrated in the surface blebs of apoptotic cells. Furthermore, their structure is altered during some types of cell death to generate structures not previously generated during development and homeostasis. This review highlights the susceptibility of SS autoantigens to undergoing such structural changes during activation of immune effector pathways, and synthesizes a model of SS incorporating these concepts. An understanding of the mechanisms responsible for activating the specific immune response in SS, and the role of specific immune effector pathways in propagating both the autoimmune response and tissue damage, is of potential therapeutic importance. Abbreviations used in this paper are: CTL, cytotoxic T-lymphocytes; ER, endoplasmic reticulum; GluR3, subunit III of the glutamate receptor; GrB, granzyme B; M3R, type III muscarinic receptor; NK cells, natural killer cells; PARP, poly(ADP-ribose)polymerase; SS, Sjögren’s syndrome; SLE, systemic lupus erythematosus; and UV, ultraviolet.
Introduction
Sjögren’s syndrome (SS) is a member of the family of systemic autoimmune diseases, in which the immune response is activated to recognize a limited variety of ubiquitously expressed autoantigens, and during which several target tissues may become damaged. Although SS is characterized primarily by chronic sialadenitis and dacryoadenitis, a variety of other tissues (including lung, nervous system, and synovial joints) may also be involved. The disease may be isolated (when it is termed ‘primary SS’), or it may occur in association with other rheumatic diseases like rheumatoid arthritis, scleroderma, or systemic lupus erythematosus (SLE) (where it is termed ‘secondary SS’). The pathogenesis of specific tissue damage in SS remains unclear, but several broad mechanisms have been proposed. These include post-infectious damage, primary salivary epithelial cell defects (including abnormal degeneration and cell death processes), chronic salivary inflammatory processes, and immune-mediated salivary damage. A currently favored pathogenic paradigm places the immune system at a central position in the genesis of ongoing tissue damage in SS. Defining the exact mechanisms (i) underlying the initial activation of the immune system in SS and (ii) responsible for the subsequent damage of target tissues remains a major priority in the disease. One of the central clues to the pathogenesis of SS comes from the observation that the immune system in SS targets a restricted and highly specific group of intracellular autoantigens, which are ubiquitously expressed in many tissues. The clustering and marked concentration of these molecules in the surface blebs of apoptotic cells, and their modification by apoptosis-specific proteolytic cleavage and/or other post-translational modifications, have focused attention on a unique apoptotic setting as the potential initiating stimulus for systemic autoimmunity in SS. The relevant immunizing event likely (i) occurs in a tissue microenvironment containing high concentrations of the targeted antigens, (ii) is pro-immune (e.g., viral infection), and (iii) allows suprathreshold concentrations of antigen with non-tolerized structure (either novel fragments, post-translational modifications, or complexes) to enter the class II antigen-processing pathway and initiate a primary immune response. Once the primary immune response to cleaved antigens has been initiated, other cleavage events (occurring in the course of homeostasis and/or damage) may drive the secondary immune response, resulting in a self-sustaining cycle of tissue damage. This review highlights the modifications of autoantigen structure that occur during different forms of cell death (particularly apoptosis), with emphasis on the salivary gland in human SS and relevant animal models. Such changes in structure in distinct microenvironments provide important insights into the initiating and propagating events in systemic autoimmune diseases, with potential therapeutic implications.
Autoantibodies: Probes of the Perturbed State
The cellular antigens targeted by a high-titer autoantibody response in systemic autoimmune diseases are a diverse group of molecules that are ubiquitously expressed, and that share no obvious features in terms of subcellular distribution, protein structure, or function (Tan, 1991). In spite of such extraordinary diversity, the specificity of the autoimmune response is remarkably predictive of disease phenotype, such that specific autoantibodies have become clinically useful diagnostically and prognostically (von Muhlen and Tan, 1995). For example, antibodies to nucleosomes are strongly associated with the SLE phenotype (Mohan et al., 1993), while antibodies to topoisomerase-1 are associated with diffuse scleroderma (Shero et al., 1986), and particularly with lung disease in such patients (Greidinger et al., 1998; Steen et al., 1988). Similarly, antibodies recognizing components of the centromere (e.g., CENP-B) are associated with the limited form of scleroderma (Moroi et al., 1980), and are predictive of digit loss in this disease (Wigley et al., 1992). Interestingly, although some autoantibody specificities (e.g., Ro/SS-A and La/SS-B) are highly enriched in patients with SS, their presence is not specific for this entity, since they are found with some frequency in patients with SLE and other systemic autoimmune diseases (Harley et al., 1992). While the targeted antigens in the different diseases do not share features that are readily apparent, there is a growing consensus that the highly specific autoantibody response to these molecules is T-cell-dependent, and that flares of disease result when this primed immune system is rechallenged with self-antigen (reviewed in Diamond et al., 1992; Burlingame et al., 1993; Radic and Weigert, 1994). Autoantibodies may therefore be surrogates which identify the primary specificities of the autoreactive T-cell response in autoimmune diseases. Since initiation of a primary T-cell response requires that a unique molecular structure (not previously generated during development of immune tolerance) be presented to the immune system in a pro-immune context, we have proposed that autoantigens likely satisfied these criteria during disease development (Rosen and Casciola-Rosen, 1999). How might such unique autoantigens be generated during disease initiation, and how might epitopes of autoantigens escape tolerance induction?
There is universal acknowledgment of the critical role of proteolysis within the endosome-lysosome pathway during the processing of antigens for presentation on MHC class II (Tortorella et al., 2000). These proteolytic events are responsible for the stereotyped and predictable outcome of the antigen-processing pathways, followed by whole protein antigens. For many antigens, antigen structure and initial antigen-processing events play a large role in the selection of determinants that predominate when loaded onto specific MHC class II molecules (dominant determinants). Since, for self-antigens, T-cell tolerance is predominantly focused on such dominant determinants (which are generated and presented at suprathreshold concentrations during natural processing of whole protein antigens), a potential exists for T-cell autoreactivity directed against ‘cryptic’ determinants (which are generated at subthreshold concentrations during normal antigen processing (reviewed in Gammon et al., 1991; Sercarz et al., 1993). Such T-cells recognizing the cryptic self never encounter their antigen during natural antigen presentation, and are therefore not tolerized. Indeed, several experimental systems have now clearly demonstrated the existence of such T-cells in vivo (reviewed in Lehmann et al., 1992; Lanzavecchia, 1995). When the structure of antigens is changed (for example, by novel autoantigen cleavage [Mamula, 1995]) or altered autoantigen processing is induced by high-affinity ligand binding (e.g., to an antibody or receptor molecule [Watts and Lanzavecchia, 1993; Salemi et al., 1995; Simitsek et al., 1995]), the hierarchy of epitopes which are efficiently loaded onto MHC class II molecules is altered, sometimes resulting in presentation of previously cryptic epitopes. We have proposed that autoantigens in systemic autoimmune diseases are structurally altered during disease initiation, thus allowing for the efficient loading of previously cryptic epitopes onto class II. This in turn results in the activation of autoreactive T-cells, which provide effective T-cell help to autoantibody-producing B-cells and probably cytotoxic lymphocytes (Casciola-Rosen et al., 1994a, 1996, 1999). High-titer autoantibodies therefore provide excellent probes of the cell biology and biochemistry of autoantigens during different clinically relevant perturbed states, to search for those circumstances in which autoantigens become clustered, concentrated, and structurally modified (Casciola-Rosen et al., 1994a,b, 1995, 1996, 1999; Casiano et al., 1996, 1998; Utz et al., 1997, 1998; Andrade et al., 1998; Odin et al., 2001). The association of particular autoantibodies with specific disease phenotypes may allow for the identification of unique modifications of autoantigen structure that occur during a pro-immune initiating event in the relevant target tissue (Andrade et al., 1998, 2001; Casciola-Rosen et al., 1999; Rosen and Casciola-Rosen, 1999).
This review highlights the susceptibility of SS autoantigens to undergoing such structural changes during immune effector pathways, and synthesizes a model of SS incorporating these concepts. Of necessity, elements of this model are speculative, but the different components are eminently testable in vitro and in vivo.
Several Intracellular Molecules are the Targets of a High-titer Autoantibody Response in SS.
Over the past several decades, numerous autoantigens have been defined in patients with SS (von Muhlen and Tan, 1995). Although there is general agreement that the primary target tissue in SS involves salivary and lacrimal epithelial cells, most of the studies performed to date have not used cells from these sources to define autoantibody specificities in this disease. The specificities defined therefore are highly enriched for molecules whose expression is ubiquitous and found in the epithelial cells used for initial screening studies. Defining specific salivary epithelial targets of the autoimmune response in SS remains a major challenge, and several recent studies have begun to ask these important questions (see below).
The ubiquitously expressed autoantigen targets in SS include the ribonucleoprotein autoantigens Ro/SS-A and La/SS-B (Harley et al., 1986), as well as several coiled-coil-containing molecules (including nuclear mitotic apparatus protein [NuMA] and members of the golgin family) (Price et al., 1984; Chan and Fritzler, 1998; Mancini et al., 2000). There have also been reports of autoantibodies recognizing α- and β-fodrin in Japanese cohorts of patients with SS, but not other autoimmune rheumatic diseases (Haneji et al., 1997; Kuwana et al., 2001). While the prevalence of such antibodies is apparently very high in Japan, it appears to be lower in cohorts in the US and Europe, and less specific for primary SS (Witte et al., 2000, and unpublished data). Antibodies to α-fodrin, however, are found with high prevalence in the NFS/sld mouse model of SS, and might provide important pathogenic insights in that model (see below). Other molecules described as autoantigens in SS include poly(ADP)ribose polymerase (PARP) (Yamanaka et al., 1987), NuMA (Price et al., 1984), Golgins (Fritzler et al., 1993, 1995; Chan and Fritzler, 1998), Ku (Yaneva and Arnett, 1989), 90-kDa nucleolar organizer region protein (NOR-90) and p80 coilin (von Muhlen and Tan, 1995), and CENP-C (Gelber et al., 2002). It is important to note that these molecules have little in common in terms of structure, subcellular distribution, and function. Furthermore, there are few data about whether antibodies directed against these targets have any direct functional effects on the salivary gland epithelium.
Several recent studies have defined autoantigens in SS that have a much more restricted expression pattern than the molecules described above. These antigens include an islet cell autoantigen (ICA), ICA-69, which is the target of autoantibodies in patients with SS, as well as in the NOD mouse model of SS (Winer et al., 2002). ICA-69 is a 69-kDa molecule expressed in pancreatic islets, brain, and salivary and lacrimal glands. Patients with SS have B-cell and T-cell reactivity directed against ICA-69, as do NOD mice, which exhibit an autoimmune sialadenitis and dacryoadenitis. Interestingly, disruption of the ICA-69 locus in NOD mice prevented lacrimal disease and greatly reduced salivary gland disease, suggesting that immunoreactivity against ICA-69 might play a role in ongoing gland damage in this model (Winer et al., 2002).
Another specificity that may be of pathogenic relevance is that directed against the type III muscarinic receptor (M3R) (Bacman et al., 1996, 1998, 2001; Robinson et al., 1998; Waterman et al., 2000). Since transduction of the autonomic signal in the salivary gland is predominantly through M3R, antibodies to this molecule would be of particular consequence through functional inhibition of salivary epithelial signaling. While most of the publications referenced above demonstrated that sera from patients with SS or NOD mice do inhibit signaling though M3R, it remains unclear whether this inhibitory activity is mediated by autoantibodies, and the precise specificity of the activity remains to be established. Defining additional salivary-specific targets of the autoantibody response in SS remains a high priority.
Sjögren’s Syndrome Autoantigens are Redistributed in Apoptotic Cells.
Cell death is among the candidate processes that may provide a source of autoantigen during the initiation and propagation of SS and other systemic autoimmune diseases (Casciola-Rosen et al., 1994a; Rosen and Casciola-Rosen, 1999). Autoantigens, which are not restricted to any specific subcellular compartment in control cells, are strikingly redistributed in apoptotic cells, such that they become clustered and concentrated within small surface blebs and apoptotic bodies (Fig.). Thus, small surface blebs (which contain fragmented rough endoplasmic reticulum [ER]) are highly enriched in 52-kDa Ro, as well as those autoantigens found within the ER lumen (e.g., calreticulin) (Rosen et al., 1995). This marked enrichment of autoantigens in small surface blebs is accompanied by their concomitant depletion from the cytosol (Casciola-Rosen et al., 1994a). Nuclear autoantigens also undergo a striking redistribution and concentration during apoptosis (Fig.). Thus, 60-kDa Ro/SS-A, La/SS-B, the snRNPs, Ku, and PARP, which normally have a diffuse nuclear distribution, initially become concentrated as a rim around the condensing chromatin in early apoptosis (Casciola-Rosen et al., 1994a, 1996). As apoptosis progresses and the nucleus becomes fragmented into multiple membrane-bound apoptotic bodies, nuclear autoantigens remain rimmed around the condensed chromatin. Such clustering of autoantigens during apoptosis has been observed in response to several additional stimuli (Rosen et al., 1995; Casciola-Rosen et al., 1999), and in numerous different cell types, including salivary epithelial cells (Ohlsson et al., 2002a). The phenomenon is not observed in cells dying by necrosis.
Several recent studies have extended these findings to demonstrate that, in addition to being clustered within apoptotic surface blebs, autoantigens also appear at the surfaces of non-permeabilized apoptotic cells (Cocca et al., 2002). This provides an important opportunity for autoantibodies to access these autoantigens, potentially activating pro-inflammatory cytokine secretion from macrophages (Manfredi et al., 1998a,b; Fadok et al., 2001), or initiating antibody-dependent cellular cytotoxicity through macrophages or NK cells. These pathways may be important in amplifying tissue damage in response to low levels of apoptotic death within the target tissue. This is particularly relevant in SS, where the absolute amount of apoptotic death of salivary gland epithelial cells appears to be quite low (Ohlsson et al., 2002b). Although there were initial reports of higher amounts of apoptotic death in SS salivary glands (Humphreys-Beher et al., 1999), such observations have not been reproduced in other studies.
While exposure of autoantigens at the surfaces of apoptotic cells may provide a mechanism for activating and amplifying immune effector mechanisms that propagate tissue damage, a critical question remains as to how the autoantibody response recognizing such clustered antigens is primarily initiated. Several mechanisms which alter the structure and expression of autoantigens during apoptosis have recently been described, and include novel proteolysis, phosphorylation, glutathiolation, transglutamination, citrullination, or formation of novel protein-protein or protein-nucleic acid complexes (Utz and Anderson, 1998; Rosen and Casciola-Rosen, 1999; Doyle and Mamula, 2002). We will use the cleavage of SS autoantigens during different forms of cell death as the paradigmatic modification with which to view the role of altered autoantigen structure in the pathogenesis of autoimmunity.
Susceptibility to Efficient Cleavage by Caspases Unifies a Subgroup of SS Autoantigens.
The clustering and concentration of autoantigens in surface blebs on apoptotic cells led us to address whether the structure of autoantigens is specifically modified during cell death, perhaps through death-specific proteolysis. We were particularly intrigued by the observation that the first proteolytic victim of the caspases discovered in apoptosis was PARP (Kaufmann et al., 1993), an autoantigen in SS and other systemic autoimmune diseases (Yamanaka et al., 1987). Proteolysis plays an important mechanistic role in the apoptotic pathway, accomplished through the specific cleavage of a limited number of downstream substrates (reviewed in Thornberry and Lazebnik, 1998; Hengartner, 2000). This apoptosis-specific proteolysis is catalyzed by a unique family of cysteine proteases (called caspases, for cysteine proteases that cleave after aspartic acid), that have an absolute requirement for aspartic acid in the substrate P1 position. Over the past several years, a significant amount of data has accumulated demonstrating that numerous SS autoantigens are cleaved by caspases during apoptosis. Relevant autoantigens include La/SS-B, GM-130 and Golgin-160, α- and β-fodrin, Ku/DNA-PKcs, NuMA, and CENP-C (Casciola-Rosen et al., 1994b, 1995, 1996, 2001; Martin et al., 1995; Casiano et al., 1996; Waterhouse et al., 1996; Haneji et al., 1997; Utz et al., 1998; Ayukawa et al., 2000; Schachna et al., 2002). Since the total number of proteins efficiently cleaved by the caspases during apoptosis appears to be restricted to several hundred molecules at most, the striking enrichment of SS autoantigens among this group of substrates is of great interest. In each case, cleavage appears to separate important functional and regulatory domains, with functional consequences. For example, cleavage of PARP removes the regulatory domain and initiates unregulated ADP-ribosylation activity (Scovassi and Poirier, 1999). Similarly, caspase-mediated cleavage of La/SS-B removes the C-terminal domain containing the nuclear localization sequence, which results in redistribution of La/SS-B from the nucleus to the cytoplasm (Ayukawa et al., 2000). It is important to note that while caspase-mediated cleavage of purified substrates in vitro is frequently highly efficient, some molecules (e.g., La/SS-B) are inefficiently cleaved in intact cells during apoptosis. Defining the immunological relevance of caspase-mediated cleavage remains an important priority (see below).
While susceptibility to efficient cleavage by a caspase is a frequent feature of several SS autoantigens, it is not a universal feature of all the autoantigens targeted in this disease. For example, caspase-mediated proteolytic cleavage of components of the frequently targeted Ro/SS-A particle has not been observed (Casciola-Rosen et al., 1996; Casiano et al., 1996). Furthermore, susceptibility to caspase-mediated cleavage does not appear to be specific to autoantigens. This imperfect, albeit striking, correlation of susceptibility to caspase cleavage and status as an autoantigen suggests that additional (and potentially partially overlapping) properties might be relevant. For example, (i) caspases may have evolved to cleave a specific regulatory motif in proteins—the association with autoantigen status may be with the presence of this protein structure rather than with cleavage during apoptosis; (ii) such structure may also be the target of additional proteases during specific forms of apoptosis or inflammation in unique tissues/microenvironments (e.g., granzyme B [GrB], matrix metalloproteases; see below); and (iii) such a regulatory motif may be subject to additional post-translational modifications either during apoptosis or during other physiologic states relevant to disease propagation (e.g., phosphorylation, glutathiolation, transglutamination, citrullination, or formation of novel protein-protein or protein-nucleic acid complexes). In this regard, it is of interest that numerous autoantigens are phosphorylated during a variety of physiologic perturbations (including apoptosis) (Utz et al., 1997). Furthermore, in several cases, recognition by autoantibodies is dependent on the phosphorylation state of the antigens (Satoh et al., 1994; Neugebauer et al., 2000).
Autoantigen clustering and cleavage occur in almost all forms of apoptosis described to date, which often occur in actively anti-inflammatory and non-immune contexts (Voll et al., 1997; Fadok et al., 1998; Hoffmann et al., 2001; Huynh et al., 2002). The marked frequency of apoptosis in normal development and homeostasis, coupled with the infrequency of systemic autoimmunity in the population, strongly suggests that only a very restricted subset of apoptotic events (e.g., those occurring in a pro-immune setting; see below), in individuals who are genetically predisposed to the generation of novel autoantigen structure (e.g., from abnormalities in the clearance and degradation of apoptotic material in tissues; see below), will initiate a self-sustaining autoimmune response.
Novel Autoantigen Fragments are Produced during Cytotoxic Lymphocyte-induced Target Cell Apoptosis.
One form of apoptosis that frequently occurs in a pro-immune setting is the death of virally infected target cells, induced by cytotoxic T-lymphocytes (CTL) and natural killer (NK) cells. Cytotoxic lymphocytes use several pathways to induce target cell apoptosis, including Fas-ligation and granule exocytosis (reviewed in Henkart, 1994; Russell and Ley, 2002; Trapani and Smyth, 2002). GrB, a serine protease found in the cytoplasmic granules of CTL and NK cells, has a substrate specificity similar to that of the caspases, in its near-absolute requirement for aspartic acid in the substrate P1 position (Poe et al., 1991). (The substrate residues immediately upstream of the cleavage site are termed P1, P2, P3, and P4 as distance increases from the scissile bond; previous studies have shown that the specificity of GrB for its substrates resides in part in the P1-P4 residues.) Although GrB also has a specificity in the P2-P4 substrate positions similar to that of the upstream activating (group III) caspases (which prefer Ileu/Val in P4 and Glu in P3; see Thornberry et al., 1997; Harris et al., 1998), there are some amino acids in the P2 and P3 positions that are preferred exclusively by GrB and not tolerated by the caspases (e.g., proline in P2, and glycine or serine in P3). Recent studies have shown that GrB plays an important role in inducing apoptotic changes in target cells during granule exocytosis-induced cytotoxicity (Shi et al., 1992; Heusel et al., 1994; Shresta et al., 1995), partly by catalyzing the cleavage and activation of several caspases (reviewed in Salvesen and Dixit, 1997; Russell and Ley, 2002). The distinguishing features of caspases and GrB are compared in Table 1. GrB also initiates caspase-independent pathways which contribute to target cell death through direct targeting of several downstream caspase substrates that mediate critical components of the apoptotic phenotype. Thus, GrB directly cleaves Bid at a site close to that utilized by caspase-8, and recruits the downstream mitochondrial pathway (Li et al., 1998; Heibein et al., 2000; Sutton et al., 2000; Alimonti et al., 2001; Pinkoski et al., 2001). The release of pro-apoptotic mediators (e.g., SMAC/Diablo) from the mitochondrial matrix into the cytoplasm also plays a critical role in deregulating the restraint imposed by ‘inhibitors of apoptosis’ proteins (IAPs) on caspase activity (Goping et al., 2003; Sutton et al., 2003). Similarly, GrB cleaves inhibitors of the caspase-activated DNAs (ICAD), allowing for activation of the caspase-activated DNAase, thereby generating the inter-nucleosomal DNA degradation that is characteristic of caspase-mediated apoptotic death (Thomas et al., 2000). We have demonstrated that the majority of autoantigens targeted across the spectrum of systemic autoimmunity are directly and efficiently cleaved by GrB, both in vitro and in cells undergoing granule-induced cytotoxicity (Andrade et al., 1998; Casciola-Rosen et al., 1999, 2001; Nagaraju et al., 2001). GrB-mediated cleavage of these substrates generates unique fragments not generated during any other form of apoptosis studied to date. Interestingly, efficient cleavage by GrB (with the generation of distinct fragments) has also been observed for several of those systemic disease autoantigens that are not cleaved by caspases. These molecules include the scleroderma autoantigens CENP-B, fibrillarin, and B23, PMS1 targeted in myositis, and the M3R targeted in Sjögren’s syndrome (Casciola-Rosen et al., 1999, 2001; Nagaraju et al., 2001). The recent demonstration that GrB cleaves subunit III of the glutamate receptor (GluR3) (an autoantigen in Rasmussen’s encephalitis) is also of significance (Gahring et al., 2001). Interestingly, the GrB cleavage site is located within the epitope targeted by antibodies in this disease. In GluR3, the GrB cleavage site faces the outside of the plasma membrane, and cleavage efficiency is markedly influenced by the glycosylation state of the receptor (Gahring et al., 2001). Thus, when GluR3 is fully glycosylated, it is relatively resistant to cleavage by GrB; the deglycosylated or non-glycosylated form of GluR3 is efficiently cleaved. The authors have proposed that generation of an ‘under’-glycosylated form of GluR3 during inflammatory states may allow for GluR3 cleavage by GrB, and the generation of previously cryptic peptide fragments (Gahring et al., 2001). Demonstrating that such circumstances occur in vivo will provide important evidence to support such a mechanism.
The striking susceptibility of many autoantigens to cleavage by GrB, together with their clustering at the same site in apoptotic cells, focuses attention on granule-induced apoptosis as a potential initiator of autoimmunity in the susceptible host. It will be extremely important to evaluate whether the immunogenicity of these novel forms of autoantigen is indeed increased over native forms of these molecules, and to demonstrate the presence of the relevant cleavage products in vivo during disease initiation and propagation. It is also possible that the susceptibility of autoantigens to cleavage by GrB reflects some unique protein conformation which has a determinative property in terms of subsequent immunogenicity. Definition of the structure of GrB cleavage sites within autoantigens, and their interactions with components of the antigen-processing pathway, is an important task.
The cytotoxic lymphocyte granule pathway has been demonstrated to be active in several circumstances in vivo. These include lymphocyte-mediated killing of virally infected target cells or cells that have undergone malignant transformation (Russell and Ley, 2002; Trapani and Smyth, 2002). A critical role for the granule pathway in down-modulation of the antigen-specific immune response has also been demonstrated (Badovinac et al., 2000). Such circumstances may have particular relevance for the initiation of systemic autoimmune diseases like Sjögren’s syndrome, and provide a paradigm within which we can understand disease initiation and propagation. For example, viral infection or transformation of salivary epithelial cells may play important roles in making these cells the targets for initial immune attack by the cytotoxic lymphocyte granule pathway. During this initial event, the granule pathway may generate altered structure of autoantigens (preferentially expressed at this site or specifically increased during this perturbation) and initiate an autoimmune response (see below).
Another important issue to consider in evaluation of the potential role of the cytotoxic lymphocyte granule pathway in the initiation of autoimmune diseases is that viral infection and transformation are encountered very frequently throughout the life of the individual, while autoimmunity is decidedly more uncommon. Numerous studies have now demonstrated that the CTL granule pathway efficiently activates downstream caspases, and directly recruits the mitochondrial pathway through cleavage of Bid (Li et al., 1998; Heibein et al., 2000; Sutton et al., 2000; Alimonti et al., 2001; Pinkoski et al., 2001). This recruitment of downstream caspase pathways is a highly amplified event, ensuring that cells die with the predominant biochemical features of the default caspase pathway under most circumstances. For example, La/SS-B, golgins, PARP, NuMA, and fodrin are all cleaved by both caspase-3 and GrB, at distinct sites. During CTL granule-induced death in normal target cells, almost all of the fragment generated is from caspase-3 activity (Andrade et al., 1998; Casciola-Rosen et al., 1999). In contrast, when endogenous caspases in the target cells are specifically inhibited, generation of the caspase-mediated fragments is abolished, and fragments directly generated by GrB are formed. This is of particular interest in SS salivary gland epithelial cells, which express high concentrations of anti-apoptotic Bcl-2 family members (Kong et al., 1998). In contrast to Bcl-2 family members, knowledge about the expression of other anti-apoptotic proteins in SS is still rudimentary. Of particular interest is the expression of the inhibitors of apoptosis (IAP) proteins, including X-IAP. It is likely that the initial generation of unique autoantigen structure occurs under such circumstances.
It is also possible that other unique structural autoantigen modifications occur during caspase-independent death pathways, and that GrB-induced fragments are only one relevant example of a more general phenomenon. For example, the lack of glutathiolation of PDC-E2 observed in cholangiocytes (in which high-level expression of Bcl-2 abrogates this modification normally observed in other cells) may generate a unique form of this primary biliary cirrhosis autoantigen that is not generated at sites of tolerance development (Odin et al., 2001).
Autoantigen Cleavage in SS: Insights from the NFS/sld Mouse Model
Over the past several years, Hayashi and colleagues have described a mouse model of SS, in which animals develop autoantibody-positive sialadenitis with functional impairment of salivary secretory function (Hayashi et al., 1996, 2000; Haneji et al., 1997; Yanagi et al., 1998). In studies to define autoantigens recognized by autoantibodies in this model, the investigators identified a 120-kDa cleavage fragment of α-fodrin as a prominent antigen in the majority of affected animals. Interestingly, this fragment co-migrated with that generated by caspase-mediated cleavage of fodrin, generated during apoptotic cell death. The specific fodrin fragment could be demonstrated in salivary glands from affected animals, but not in those from controls. When the fragment was used to tolerize animals early in life, development of sialadenitis was prevented, demonstrating a role for an immune response to cleaved fodrin in the initiation and propagation of ongoing salivary gland pathology in these animals (Saegusa et al., 2002). Similarly, prevention of caspase activity in vivo partially ameliorates development of salivary gland dysfunction in affected animals (Saegusa et al., 2002). Although a similar α-fodrin fragment is generated by caspases, there is no direct evidence that caspases are responsible for generating this fragment in vivo. Indeed, additional proteases active in the salivary gland may play important roles (see below).
Although the prevalence of anti-fodrin autoantibodies in human SS patients and the presence of fodrin cleavage fragments in SS salivary glands remain controversial, the Hayashi studies have important implications. First, they suggest that autoantigen cleavage occurs in affected tissues in the mouse model. Second, they demonstrate that such cleavage may play an important role in the selection of autoantigens for an autoimmune response, and in generating ongoing tissue damage. It is of interest that, at the hinge region between the N- and C-terminal domains, fodrin is highly susceptible to cleavage by several other proteases, including calpains (Siman et al., 1984) and GrB (Nagaraju et al., 2001). Defining the physiologically critical protease responsible for the initial cleavage of fodrin in this model is important.
Unique Protease Expression in the SS Salivary Gland: Potential for Generation of Altered Autoantigen Structure in the Unique Disease Microenvironment
The studies described above have focused on a very limited group of modifying forces that might be important for altering autoantigen structure in salivary gland epithelial cells. It must be remembered that the inflammatory microenvironment contains numerous other effector components which might change levels of antigen expression and antigen conformation, as well as protease expression, in resident and immigrant cells. For example, a recent study from Baum’s group demonstrated that matrix metalloprotease expression is markedly up-regulated in salivary epithelial cells in response to cytokine exposure (Wu et al., 1997). Immigrant inflammatory cells also bring with them a complement of proteases and other activities which might alter the structure of autoantigens. Systematically addressing the effects of such forces on SS-specific autoantigen structure is an important priority.
Model of SS
The studies presented above have focused attention on unique pro-immune events in specific microenvironments as processes that generate unique autoantigen structure, and thereby initiate a self-directed immune response in the systemic autoimmune diseases. We propose that, in the genetically susceptible individual, the confluence of several forces allows for the effective initiation of the primary immune response (Table 2). Such forces generate suprathreshold concentrations of antigens with structure not previously tolerized by the host, and facilitate effective capture of such antigens by antigen-presenting cells, and presentation to T-cells. The molecules targeted are unified by their susceptibility to structural modification during the perturbing process, likely revealing previously cryptic structure. Once primary immunization has occurred, the effector pathways activated by the primed immune system include several which themselves may generate an ongoing supply of modified antigen (e.g., cytokine effects, novel protease expression), driving further immune response and effector pathways. Our understanding of the pathways relevant to disease initiation and propagation will provide a rational basis for therapy for a disease which places an extremely high burden on those who suffer from it, and in which targeted and effective therapies remain elusive.
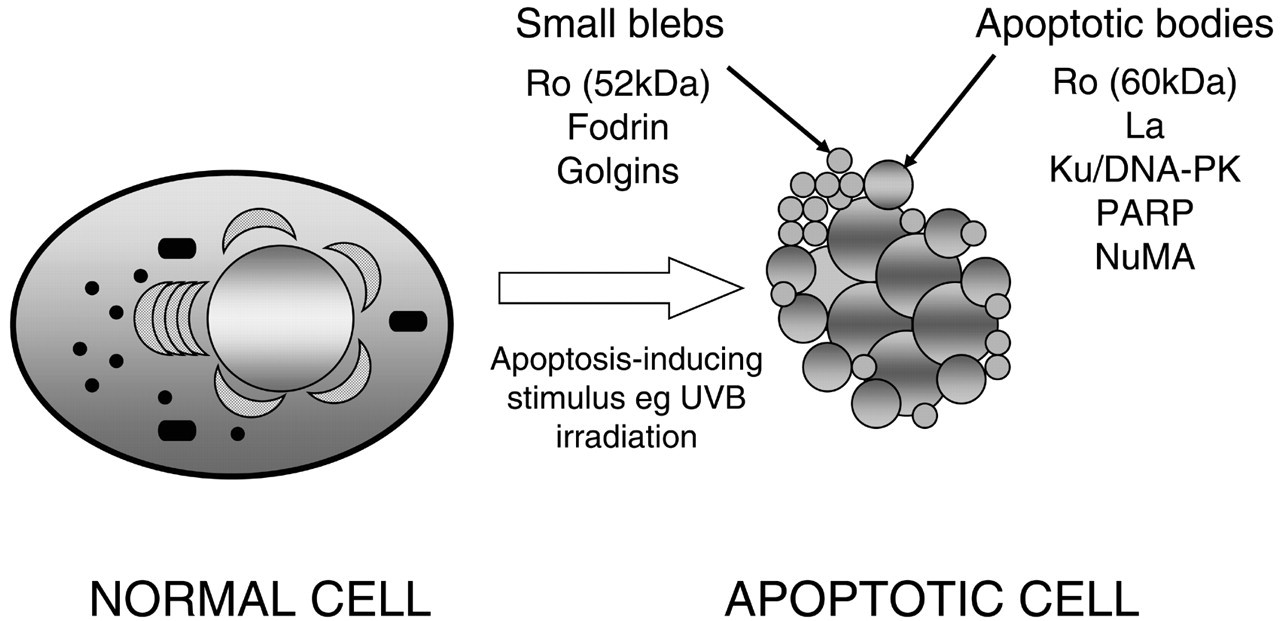
Autoantigens cluster in unique subcellular structures generated during apoptosis. A normal and an apoptotic cell are shown in this Fig. (
Footnotes
Acknowledgements
This work was supported by National Institutes of Health (NIH) grants AR44684 (LCR) and R37 DE12354 (AR; MERIT Award), by the Edmund C. Lynch Fellowship in Arthritis Research in honor of Vernon Lynch, and by an Arthritis Foundation Maryland Chapter MARRC Grant. A. Rosen is supported by a Burroughs Wellcome Fund Translational Research Award.