
Abstract
It is generally accepted that the nervous system contributes to the pathophysiology of peripheral inflammation, and a neurogenic component has been implicated in many inflammatory diseases, including periodontitis. Neurogenic inflammation should be regarded as a protective mechanism, which forms the first line of defense and protects tissue integrity. However, severe or prolonged noxious stimulation may result in the inflammatory response mediating injury rather than facilitating repair. This review focuses on the accumulating evidence suggesting that neuropeptides have a pivotal role in the complex cascade of chemical activity associated with periodontal inflammation. An overview of neuropeptide synthesis and release introduces the role of neuropeptides and their interactions with other inflammatory factors, which ultimately lead to neurogenic inflammation. The biological effects of the neuropeptides substance P (SP), calcitonin gene-related peptide (CGRP), vasoactive intestinal polypeptide (VIP), and neuropeptide Y (NPY) are summarized, and evidence for their involvement in the localized inflammatory lesions which characterize periodontitis is presented. In this context, the role of CGRP in bone metabolism is described in more detail. Recent research highlighting the role of the nervous system in suppressing pain and inflammation is also discussed.
(1) Introduction
Inflammation represents the response of vascular tissues to insult or injury. It is usually protective but, if the causative agent persists, can become chronic with associated tissue damage. Considerable research is currently focusing on how inflammation is set in motion and ultimately controlled, with a view to the development of novel therapeutic interventions. The magnitude of the inflammatory response is crucial, since an insufficient response can lead to infection, whereas an excessive one can cause morbidity, e.g., due to bystander damage in diseases such as rheumatoid arthritis. The magnitude of the inflammatory responses to plaque micro-organisms, particularly their lipopolysaccharide (LPS) component, is crucial in determining the extent and duration of gingivitis and periodontitis. The inflammatory response to LPS is the result of a complex series of interactions between soluble factors and cells whose functions need to be precisely regulated to defend the body and promote healing. In recent years, the nervous system has been identified as a critical regulator of inflammation (Tracey, 2002). This review will therefore focus on the role of the nervous system in the inflammatory process and, in particular, the contribution of the nervous system to periodontal inflammation.
More than a century ago, Bayliss (1901) made the first observations that activation of dorsal root ganglia neurons resulted in peripheral vasodilation. He concluded that neurons not only conducted information to the spinal cord (afferent conduction) but also served to relay information from the spinal cord to the periphery (efferent conduction). Strong support for the above description of the axon reflex was later obtained in human skin. The so-called classic ‘triple response’ observed in response to injury was characterized by Lewis (1927) to be comprised of a line (due to the reaction of the capillaries to the mechanical stimulus), a spreading flare observed up to several centimeters around the injury (due to arteriolar dilation and increased blood flow), and a subsequent wheal (due to increased capillary permeability and local edema). The flare and wheal were considered to be the consequence of a sensory-mediated axon reflex, thought to be a signaling device to warn adjacent tissues of trauma. Several years later, von Euler and Gaddum (1931) isolated a compound, capable of decreasing blood pressure, which they called substance P (SP). It was not until 1953 that Lembeck postulated that SP could be a sensory neurotransmitter, a theory vigorously tested 20 years later when synthetic SP became available. The vasodilatory response to SP and the verification of SP as a neurotransmitter confirmed the suggestion by Dale (1935) that the chemical transmitter of the axon-reflex vasodilation might be identical to the sensory transmitter at the spinal cord synapses, because they were both released from the same neuron, one from peripheral terminals and one from central terminals. It is now assumed that axon reflexes involving sensory nerves occur in most tissues of the body and are central to the proposed pro-inflammatory role of neuropeptides in disease. Many studies have demonstrated a role for sensory neurons in vascular aspects of inflammation, and the term ‘neurogenic inflammation’ has been coined to define the contribution of the nervous system to local inflammatory responses (Jansco et al., 1967).
(2) Overview of Neuropeptides
All nerve cells or neurons perform the same basic tasks. Every neuron generates an electric impulse in response to a chemical or mechanical stimulus, conducts the impulse through its elongated cell structure, and, at its terminal, translates the electrical activity into a chemical signal. The active chemical, called a neurotransmitter, crosses the synapse to the membrane of the next neuron in the neuronal circuit. There the neurotransmitter interacts with specific receptors and generates a new electrical impulse which continues the signal. Alternatively, the neurotransmitter can be liberated into the extracellular fluid, where it may have paracrine function if it acts locally through receptors on other neurons or immune cells in the immediate neighborhood.
Neuropeptides are simply peptide neurotransmitters. It is implicit that for a peptide to be defined as a neuropeptide, it is synthesized and released from neurons and has biological actions mediated via extracellular receptors on target cells (Hoyle, 1995). Neuropeptides represent the largest class of transmitter substances currently known, mediating extraordinarily diverse functions. They have a long phylogenetic history and are highly conserved, indicating an important evolutionary role (Holmgren and Jensen, 2001). In vertebrates, neuropeptides are present in every division of the nervous system. They are found in autonomic pre- and post-ganglionic nerves, primary sensory neurons, somato-sensory neurons, and neurons throughout the central nervous system (CNS). It would be impossible to examine all aspects of neuropeptide research in a review of this size. However, based on their occurrence and physiological roles in vertebrates, some features of the most prominent members of the neuropeptide family of neurotransmitters are summarized below.
(A) SP and the tachykinins
The vasoactive material extracted into a powder by von Euler and Gaddum (1931) was named substance ‘P’ for ‘powder’. SP is an 11-amino-acid peptide believed to be identical in all mammalian species. SP is a member of the tachykinin family of neuropeptides, also known as the neurokinins. In addition to SP, two other tachykinins are known to exist, neurokinin A (NKA) and neurokinin B (NKB). SP and NKA are derived from the preprotachykinin A gene as a result of alternative splicing of a common transcription product (Nawa et al., 1984). The preprotachykinin A gene has been shown to be induced by cytokines, glucocorticoids, neuropeptides, and LPS, although the mechanism remains unclear (Rameshwar, 1997). SP and NKA have been intensely studied and tend to have a similar distribution, distinct from that of NKB (Helke et al., 1991). SP and NKA are present in peripheral nerves, including enteric neurons and capsaicin-sensitive primary afferent neurons; however, NKB is not expressed in detectable amounts in most peripheral tissues and will not be dealt with further in this review.
(B) Calcitonin gene-related peptide
Calcitonin gene-related peptide (CGRP) was originally discovered in 1982, when Amara and colleagues showed that alternative RNA processing of the calcitonin gene generated mRNAs encoding a peptide they named CGRP. CGRP is a 37-amino-acid peptide with slight differences in sequence homology between different mammalian species. CGRP is widely distributed throughout the central and peripheral nervous systems and is found in particularly high levels in sensory nerves. CGRP has potent vasodilator activity (Brain et al., 1985) and is frequently co-localized with SP. Indeed, SP has been shown to regulate the vasodilator activity of CGRP (Brain and Williams, 1988), suggesting that there is an important functional significance to this co-localization.
(C) Vasoactive intestinal polypeptide
Vasoactive intestinal peptide (VIP) is a 28-amino-acid peptide originally isolated from pig intestinal extracts (Said and Mutt, 1970) and later ‘rediscovered’ in the central and peripheral nervous systems. It is now known that VIP is a ubiquitous peptide, with a wide profile of physiological actions, including relaxation in smooth-muscle cells and salivary secretion. In the last decade, VIP has been identified as an important immunomodulatory peptide (Bellinger et al., 1996), capable of regulating the production of both pro- and anti-inflammatory mediators (Pozo et al., 2000; Ganea and Delgado, 2002).
(D) Neuropeptide Y
Neuropeptide Y (NPY), a 36-amino-acid peptide originally isolated from porcine brain (Tatemoto, 1982), derives its name from its N- and C-terminal tyrosines (Y). There is complete homology of NPY in the human, rat, rabbit, and guinea pig (O’Hare et al., 1988). Both immunohistochemistry and radioimmunoassay have revealed the widespread occurrence of NPY throughout central and peripheral nervous systems, where it has been shown to have potent vasoconstrictor activity (Lundberg et al., 1985).
(E) Synthesis of neuropeptides
Neuropeptides are derived directly from transcripts of nuclear genes and are therefore synthesized on ribosomes in the perikaryon of neurons prior to axonal transport in both central and peripheral directions. With up to four times as much SP transported to peripheral branches of primary sensory neurons compared with the dorsal root (Harmar and Keen, 1982), considerable quantities of neuropeptides are therefore stored peripherally for subsequent release. Although the neuronal cell body or perikaryon is presently considered to be the sole site of ribosomal neuropeptide synthesis within the neuron, neuropeptide-encoding mRNA has been found within the axonal compartment (Jirikowski et al., 1990). The possibility of protein synthesizing machinery within axons has recently been acknowledged (Giuditta et al., 2002); however, in the light of current evidence, axonal synthesis has yet to be widely accepted by the scientific community.
Like all proteins and polypeptides, mature neuropeptides are produced by cleavage from large precursor prepropeptide molecules. The ‘pre’ (signal) is cleaved off in the endoplasmic reticulum to produce a pro-peptide (inactive precursor). The pro-peptide may contain one or more neuropeptide sequences, and, depending on the complement of processing enzymes (within the Golgi or secretory granules), the cleavage of biologically active neuropeptides may vary. This method of synthesis greatly increases the diversity of neuropeptides available for synaptic or paracrine signaling and offers some explanation for the biological expense of synthesizing large inactive precursors. The final stage of processing to biologically active neuropeptides involves post-translational modifications, which may include C-terminal amidation, N-terminal acetylation, cyclization of glutamate to pyroglutamate, disulphide bond formation, glycosylation, phosphorylation, or sulphation (Bradford, 1985). The biologically active neuropeptides are stored in so-called large (> 70 nm) dense core vesicles (LDCVs) in the synaptic cleft prior to release. The synthesis of neuropeptides is therefore a complex process, distinctly different from that of low-molecular-weight (LMW) neurotransmitters (Stahl, 1999). LMW neurotransmitters such as acetylcholine, norepinephrine, and γ-aminobutyric acid (GABA) are synthesized from dietary sources by one or two intracellular enzymatic steps. This synthesis occurs in the cytosol of the nerve terminal, where the transmitter is incorporated into small clear synaptic vesicles for storage and release.
(F) Release of neuropeptides
Neuropeptides are released from LDCVs in the synaptic cleft in response to calcium-dependent depolarization. The stimuli for the release of neuropeptides are generally of higher frequency and of longer duration than those required for the exocytosis of LMW transmitters (Burgoyne and Morgan, 1995). Once released, neuropeptides may have direct post-synaptic actions, but more often than not, they have modulator functions at post-synaptic sites. Such modulatory functions may include amplification or attenuation of LMW neurotransmitter actions (Zukowska-Grojec and Wahlestedt, 1993).
Neuropeptide actions following release, however, are not limited to only post-synaptic neurotransmitter/neuromodulator roles; in addition, neuropeptides may also have paracrine actions. Many neuropeptides act on non-neuronal targets (paracrine action) after diffusing through the local microenvironment. Functional neuropeptide receptors for SP (Hartung et al., 1986) and CGRP (McGillis et al., 1991) are present on immune cells, suggesting that the paracrine action of neuropeptides has an important immunomodulatory role. Furthermore, peptides identical to those extracted from the nervous system can, under pathological conditions, be synthesized and released from non-neuronal inflammatory cells (Metwali et al., 1994). Neuropeptides may act as neurotransmitters or neuromodulators at one synapse but have paracrine-like actions at other local cellular sites bearing their receptors. Therefore, we should not attempt to classify the individual neuropeptide but rather the action of the neuropeptide as transmitter/modulator/paracrine, depending upon its unique microenvironment.
(G) Neuropeptide co-transmission
The concept that each nerve cell made and released only one neurotransmitter at all its branches and terminals stemmed from Dale’s principle (Dale, 1935), although recent interpretation of Dale’s research suggests that this dogma was not his intention (Strata and Harvey, 1999). The neurochemical designation of a neuron based on its transmitter, however, was considered an important aspect of the neuronal phenotype. Accordingly, neurons were designated as cholinergic (acetylcholine as neurotransmitter), adrenergic (adrenaline), peptidergic (neuropeptide), and so on. This nomenclature is still used, although it tends to suggest, erroneously, neurotransmitter exclusivity. It is common to find more than one neuropeptide in a given population of neurons—for example, 80% of SP-containing nociceptive fibers also contain CGRP (Lee et al., 1985). Furthermore, many neurons release more than one neurotransmitter type, typically one or more neuropeptides and a LMW neurotransmitter, in a phenomenon known as co-transmission. This is particularly the case in the enteric nervous system, where as many as six different neuropeptides can co-exist with acetylcholine in one neuron (Steele and Costa, 1990). These neurons commonly contain large vesicles that contain both neuropeptide(s) and a LMW neurotransmitter together with small vesicles that contain only a LMW neurotransmitter. After release of both types of neurotransmitter, the neuropeptides may modulate the action of the classic LMW neurotransmitter or, alternatively, may signal to a separate group of post-synaptic cells. Thus, neurons may release a cocktail of molecules, including neuropeptides and classic LMW neurotransmitters, providing a spectrum of biological activity, which can be altered by changes in any one of the constituents (Lundberg and Hokfelt, 1983).
(H) Receptors
Neuropeptides cannot cross cell membranes, and therefore, to elicit a post-synaptic or target cell response, they must interact with a receptor. Approximately 80% of neuropeptide receptors are G-protein-coupled receptors (GPCRs), the most commonly used signal transduction system in the animal kingdom. In humans, it is estimated that approximately 1,000 distinct integral membrane GPCRs direct cellular responses to a wide variety of chemical transmitters, including neuropeptides. Neuropeptides bind in a reversible manner to their receptors, resulting in interactions with heterodimeric G-proteins and subsequent stimulation of adenylate cyclase and cAMP formation. In this signaling system, cAMP acts as a second messenger, subsequently activating cAMP-dependent kinase. By altering protein conformation, cAMP-dependent kinase ultimately regulates ion channels, enzyme activity, and/or structural proteins (for review, see Bouvier, 2001).
The fate of the GPCR following ligand binding is also important, because it determines resensitization of the target cell to the neuropeptide, which may be an important determinant of any chronic inflammatory process. Receptor fate has been studied in detail for the SP neurokinin-1 (NK-1) receptor interaction. After SP binds to the NK-1 receptor and elicits a signal, the SP-NK-1 receptor complex is internalized. Once inside the perinuclear compartment, endosomal acidification induces the dissociation of SP, which is targeted to lysosomes for degradation, and the NK-1 receptor is recycled to the plasma membrane. The uptake of SP by target cells occurs only after receptor stimulation, and so it does not regulate the concentration of neuropeptide in the extracellular fluid. SP-induced endocytosis does, however, deplete the plasma membrane of high-affinity receptors, but resensitization of cells to SP is believed to require only recycling of the receptor and not new receptor synthesis (DeFea et al., 2000). The endocytotic pathways of many neuropeptides remain to be studied in detail, so the extent of intracellular receptor sorting and recycling is yet to be determined.
(I) Fate of neuropeptides
After release, neuropeptides are metabolized by peptidases, which terminate their activity and therefore play a central role in modulating the levels of these biologically active chemicals. It is believed that the metabolic fragmentation of neuropeptides is non-specific compared with the highly selective breakdown of classic neurotransmitters (Turner, 1986). The tissue or body fluid into which the neuropeptide is released has an important regulatory influence on peptide hydrolysis, depending on local enzyme abundance and/or activity. The enzymes involved in neuropeptide degradation include: endopeptidases, which cleave peptide bonds between non-terminal amino acids; exopeptidases, which cleave amino acids from either the N- or C-terminus of the peptide; aminopeptidases, which remove non-amidated N-terminal amino acids; and carboxypeptidases, which cleave non-amidated C-terminal amino acids. It is believed that many neuropeptides are degraded by several of these enzymes in contrast to LMW neurotransmitters, each of which undergoes highly specific degradation by a single enzyme (Turner, 1986). Furthermore, neuropeptides are degraded more slowly than LMW neurotransmitters, a property that may account for the more prolonged and widespread effects of neuropeptides.
The metabolism of SP has been studied in detail and serves to demonstrate the generally accepted route of neuropeptide inactivation. SP is metabolized by the neutral endopeptidase (NEP) E.C.3.4.24.11 to the fragments SP1–6 and SP7–11 (Erdos and Skidgel, 1989). These fragments are then further degraded by amino- and carboxy-peptidases to biologically inactive molecules. The metabolism of CGRP in gingival crevicular fluid (GCF) has been studied in our laboratory (Lundy et al., 2000a) and may have an important role in the pathogenesis of periodontitis.
(J) Neuropeptides compared with low-molecular-weight neurotransmitters
The synthesis, release, and fate of neuropeptides differ in several important respects from those of LMW neurotransmitters. Neuropeptides have extremely potent effects and are present in very much smaller quantities than LMW neurotransmitters. For example, the neuropeptide concentration in the human brain is of the order of picomoles per gram of tissue, which is perhaps 100 to 1000 times less than the concentration of LMW neurotransmitters (Bradford, 1986). The rate of synthesis of neuropeptides is a relatively slow process because it is regulated by signaling events in the cell body, in contrast to the simple requirement for the re-uptake of LMW neurotransmitters by the nerve terminal without de novo synthesis. The binding of neuropeptides to their receptors occurs at much lower concentrations (10−8 to 10−10 M) than the binding of LMW neurotransmitters such as acetylcholine (10−5 to 10−6 M). Furthermore, since neuropeptide receptors act indirectly, through intracellular pathways, they can provide tremendous amplification. Although the release of neuropeptides requires more intense stimulation, they tend to be degraded more slowly than LMW neurotransmitters, and, as a consequence, significantly fewer neuropeptide molecules are needed to influence a post-synaptic target. These demands can therefore be met by a supply of molecules emanating from the cell body.
(3) Activation of Sensory Nerves
Despite our knowledge of the sensory innervation of periodontal tissues (Sinnatamby, 1999), much of the detail concerning the chemical nerve fiber subtypes innervating periodontal tissues remains to be determined. As a result, the specific contributions of various gingival and periodontal sensory nerves to neurogenic inflammation are yet to be fully elucidated. The difficulty in assigning specific roles to subclasses of sensory fibers within the periodontium is exacerbated by the fact that sensory nerves exhibit a high degree of plasticity, which may result in changes in neurochemistry, or phenotypic switching to reflect specific micro-environmental defensive or protective roles. Although the exact contributions of the subclasses of sensory nerves remain to be defined, much more is known about the chemicals or conditions that generally activate these nerves, thereby regulating neurotransmitter release.
A great many substances have been reported to activate sensory neurons, including capsaicin, heat, protons, bradykinin, and tryptase. In all cases, the noxious stimulus triggers neuropeptide release from a sensory nerve terminal; however, the exact mechanism varies, depending on the stimulus. Capsaicin, heat, and protons activate the recently cloned vanilloid receptor (VR1), resulting in the opening of ligand-gated cation channels, allowing calcium to enter the sensory nerve cell (Caterina et al., 1997). The resulting depolarization activates voltage-gated calcium channels, allowing for further calcium entry. The vanilloid receptor family, which is comprise of VR1 and other related proteins, forms a subgroup of the transient receptor potential vanilloid (TRPV) family of ion channels (Gunthorpe et al., 2002). Within the TRPV family, VR1 is distinguished as the only channel activated by vanilloids such as capsaicin. The VR1 receptor has recently been shown to be up-regulated in inflammatory bowel disease, suggesting a possible role for this receptor in sustaining the chronic nature of the disease (Yiangou et al., 2001). In contrast, other inflammatory mediators such as bradykinin and prostaglandins act via specific receptors on sensory nerves. Bradykinin has been shown to bind to B2 receptors on sensory neurons, resulting in the activation of phospholipase C and ultimately the release of calcium from intracellular stores, thereby triggering neuropeptide exocytosis (Burgess et al., 1989). The pro-inflammatory prostaglandins E2 and I2 (PGE2 and PGI2) have been shown to bind to specific receptors on sensory nerves, where they lower the firing threshold (Schaible and Schmidt, 1988). Recent work has demonstrated that bradykinin and PGE2 have a positive interaction in evoking CGRP release from bovine dental pulp (Goodis et al., 2000). Although other inflammatory mediators, such as the cytokines, do not induce neuropeptide release directly in vitro, cytokines can be expected to sensitize receptors, thereby increasing the capsaicin-evoked release of neuropeptides, possibly via receptor-associated kinases and phosphorylation of ion channels (Opree and Kress, 2000).
The recent discovery of a new class of protease-activated receptors (PARs) has shed light on the mechanism of enzyme-mediated sensory nerve activation and subsequent neuropeptide release. Research into the functionality of these receptors has been driven by the belief that PARs (especially PAR-2) have a particularly important role in disease states associated with chronic inflammation (Vergnolle, 1999). PAR-2 is co-expressed with SP and CGRP on sensory nerves, where it is believed to mediate neurogenic inflammation and may also have a role in nociception (Steinhoff et al., 2000; Fiorucci and Distrutti, 2002). PARs are characterized by a novel mechanism of receptor triggering in which proteolytic cleavage of the N-terminus subsequently activates the receptor. Much remains to be learned about the endogenous sources of the enzymes that activate PARs. However, the observation that tryptase, which constitutes the major protein released during human mast cell degranulation, can cleave PAR-2 in vitro suggests one potential source. The close spatial association observed between mast cells and nerves (Dimitriadou et al., 1994) would tend to support an important role for mast cell tryptase in activating PAR-2 on neurons, thus mediating the release of SP and CGRP and resulting in neurogenic inflammation (Vergnolle et al., 2001). In the light of current evidence, it is tempting to speculate that PAR-2 activation of sensory nerves may have a potentially important role in neurogenic inflammation associated with periodontal disease, particularly since PAR-2 could be activated by tryptase, which is known to be present in the local environment (Kennett et al., 1997). In fact, SP-immunoreactive nerve fibers have been found in the vicinity of tryptase-positive mast cells in human periapical granulomas (Kabashima et al., 2002). Furthermore, tryptase, which has no known endogenous inhibitor, has the potential to act inappropriately on PAR-2 as a chronic mediator of neurogenic inflammation (Fig.). It is possible that the typically chronic nature of the inflammation associated with periodontitis could be sustained by such a mechanism. However, the gingival crevice contains a plethora of inflammatory cells and enzymes, and the characteristic infiltration of neutrophils in response to dental plaque accumulation may implicate the neutrophil enzyme, proteinase 3, in gingival and periodontal inflammation. Proteinase 3 has recently been shown to activate PAR-2 on oral epithelial cells and induce the production of the pro-inflammatory cytokine IL-8 (Uehara et al., 2002). From the limited information currently available, it would appear that therapy aimed at controlling PAR-2-activating proteases such as tryptase and/or proteinase 3 could be of potential benefit in regulating periodontal disease.
(4) Non-neuropeptide Modulators of Neurogenic Inflammation
The neurogenic inflammatory response is complex and cannot simply be regarded as a series of neuronal events occurring in isolation. Indeed, it is known that the initiation and sustenance of neurogenic inflammation depend on a variety of factors present in the local environment.
(A) Cytokines and suppressors of cytokine signaling
Cytokine-neuropeptide interactions are bidirectional—that is, cytokines and other products of the immune cells can modulate the action, differentiation, and survival of neuronal cells, while neuropeptides released from neurons play pivotal roles in influencing the immune response. For example, cytokines have been shown to regulate neuropeptide expression, notably the expression of SP in sympathetic neurons (Kessler et al., 1993). Some but not all of this stimulation is mediated by the neuropoietic cytokine leukemia inhibitory factor (LIF). Furthermore, cytokines have been shown to have a role in the LPS-induced facilitation of CGRP release from capsaicin-sensitive nerves (Hua et al., 1996) and to enhance capsaicin-induced vasodilation in rat skin (Herbert et al., 1994). Neuropeptides not only enhance cytokine actions—for example, CGRP enhances IL-1-induced neutrophil accumulation (Buckley et al., 1991)—but can also induce T-cell cytokine secretion (Levite, 1998).
The pro- and anti-inflammatory actions of cytokines in inflammation have been studied in detail (see review by Hanada and Yoshimura, 2002). Historically, LPS has been associated with potent activation of the inflammatory response through the induction of cytokine production. However, it was not until the identification of the Toll-like receptors (TLRs) that specific receptors were linked to this response. Different TLRs recognize distinct microbial features, and although LPS generally activates TLR4, there is some evidence to suggest that LPS from Porphyromonas gingivalis can also activate TLR2 (Medzhitov, 2001). As sentinels of microbial invasion, TLRs are found at sites of host-microbe interaction. To date, a wide variety of cell types—including intestinal epithelial cells, dermal endothelial cells, and peripheral blood leukocytes—has been shown to express TLRs. It is currently thought that varied expression of TLRs together with differential responsiveness to TLR ligands allows for a specific type of cytokine response, depending on the unique microenvironment (Krutzik et al., 2001). Signaling pathways from the TLRs ultimately result in activation of mitogen-activated protein kinase and translocation of the transcription factor NF-κB, which regulates the activity of the genes that produce cytokines (Baldwin, 2001). Therefore, LPS acting through TLRs has an indirect effect on neurogenic inflammation by stimulating the production of pro-inflammatory cytokines, which are neuromodulators capable of directly influencing neuropeptides (Fig.).
LPS signalling via TLRs has been identified as a pivotal mechanism for cytokine production; however, less is known about the inactivation of such signal transduction pathways. Recently, a family of proteins, known as suppressors of cytokine signaling (SOCS), has been identified as inhibitors of the actions of cytokines (Auernhammer and Melmed, 2001). It has been shown that LPS induces SOCS-1 and SOCS-3 expression, and it has been postulated that such expression can be induced after the triggering of TLR signal pathways as part of the innate immune response (Dalpke et al., 2001). If further experimental data confirm that LPS activation of TLRs and, subsequently, of inflammatory cells is counter-regulated by LPS activation of SOCS, then this would suggest substantial cross-talk between signaling pathways within cells, highlighting the complex nature of the neuroimmune regulatory responses to LPS.
(B) Nerve growth factor
The role of neurotrophins such as nerve growth factor (NGF) in the development and maintenance of peripheral sympathetic and nociceptive sensory neurons is well-established. NGF controls the survival and phenotype of immature neurons and may also be important for the maintenance of normal phenotype in adult sensory neurons (Lindsay and Harmar, 1989). During prolonged inflammatory processes, NGF has been shown to be responsible for increased synthesis of SP and CGRP in the dorsal root ganglion (Donnerer et al., 1992). More recently, it has been shown that NGF directly regulates the synthesis of CGRP in B-cells as it does in sensory neurons (Bracci-Laudiero et al., 2002). The synthesis of other neuropeptides, however, such as VIP and NPY, appears to be partly inhibited by neurotrophins (Verge et al., 1995).
(C) Bradykinin
In the course of inflammation, ubiquitous kininogens are degraded to form kinins, including bradykinin (BK), which together are important mediators of inflammation. In addition to direct activation and sensitization of nociceptors, there is substantial evidence that kinins are pro-inflammatory, leading to vasodilation, plasma extravasation, and the release of other inflammatory mediators, notably the neuropeptides SP and CGRP (Geppetti, 1993).
(D) Nitric oxide
The unconventional gaseous neurotransmitter nitric oxide (NO) is also believed to have a role in neurogenic inflammation. It has been suggested that NO may act pre-junctionally or within peripheral neurones to mediate the release of neuropeptides during neurogenic inflammation within the skin microvasculature (Hughes and Brain, 1994; Kajekar et al., 1995). The vasodilation response characteristic of neurogenic inflammation has been shown to require the presence of endothelium and is linked to the production of NO (Lippe et al., 1993). Furthermore, angiogenesis promoted by SP has been reported to be dependent on the constitutive activation of NO synthase in vascular endothelium, leading to the generation of NO (Ziche et al., 1994).
(5) Effects of Neuropeptides
Sensory neuropeptides play important roles in neurogenic inflammation, including vasodilation, plasma extravasation, and recruitment of immune cells; however, a more extensive function for neuropeptides in the regulation of immune cell activity has also been proposed. During inflammation, there is a sprouting of peptidergic peripheral fibers and an increased content of neuropeptides (Kvinnsland and Heyeraas, 1992; Byers and Taylor, 1993; Awawdeh et al., 2002a). Peptide-containing nerve fibers are often seen existing close to immune cells, particularly macrophages (Toriya et al., 1997) and mast cells (Kabashima et al., 2002). Furthermore, the identification of functional neuropeptide receptors on immune cells (Hartung et al., 1986; McGillis et al., 1991) suggests a role for neuropeptides in neuroimmunomodulation.
(A) SP and NKA
SP and NKA are members of the tachykinin (tachy-swift) neuropeptide family and as such evoke rapid responses upon release. They exert a wide variety of biological actions and are intimately linked with neurogenic inflammation, which has been shown to have a dose-dependent relationship with the levels of SP and/or NKA (Cao et al., 1998). SP causes vasodilation by acting directly on smooth-muscle cells and indirectly by stimulating histamine release from mast cells in a concentration-dependent manner. Structure-activity relationship studies have shown that both the N-terminal residue and the hydrophobic C-terminal of SP play important roles in the histamine release mechanism (Shibata et al., 1985). Increased microvascular permeability, edema formation, and subsequent plasma protein extravasation are prominent peripheral effects of the tachykinins, underlying their powerful pro-inflammatory properties. The edema induced by SP is primarily due to increased vascular permeability mediated through its action on NK-I receptors situated on post-capillary venule endothelial cells (Lembeck et al., 1992). The SP-induced contraction of endothelial cells and subsequent plasma extravasation allow substances, such as bradykinin and histamine, to gain access to the site of injury and to afferent nerve terminals. SP also interacts with other neurotransmitters—indeed, the characteristic edema formation mediated by SP has been shown to be modulated by NO (Hughes et al., 1990).
Exogenously applied SP at physiological concentrations in the range 10−11 to 10−13 M augments IL-2 production (Rameshwar et al., 1993) and T-lymphocyte proliferation both in vitro and in vivo (Scicchitano et al., 1988; Nio et al., 1993). However, in addition to an involvement in the recruitment and stimulation of inflammatory and immune cells, SP is also synthesized extraneuronally by such cells. Peripheral extraneuronal sources of SP include monocytes, dendritic cells, eosinophils, and mast cells. Human T-lymphocytes contain preprotachykinin mRNA, encoding SP, and produce endogenous SP (Lai et al., 1998). Furthermore, activation with LPS in vitro produced a marked increase in SP expression by mononuclear phagocytes and dendritic cells (Lambrecht et al., 1999). Most immune cells producing SP also express its receptor (Lai et al., 1998). This led to the hypothesis that SP not only acts as a mediator of the crosstalk between the nervous and immune systems but also acts independently of sensory nerves in a paracrine and/or autocrine fashion to mediate direct interactions between immune cells.
(B) CGRP
Although CGRP is released simultaneously with SP and is a potent vasodilator (Brain et al., 1985; de Hoon et al., 2003), emerging roles for CGRP suggest that it has immunosuppressive effects and may be considered to down-regulate the inflammatory response (Taylor et al., 1998). In contrast to SP, CGRP has been shown to suppress IL-2 production and the proliferation of murine T-cells (Wang et al., 1992). CGRP inhibits the ability of macrophages to produce hydrogen peroxide in response to interferon gamma, to present antigen (Nong et al., 1989), and to differentiate (Owan and Ibaraki, 1994). CGRP can also directly influence the cytokine profile of unstimulated Th clones, inducing atypical cytokine secretion (Levite, 1998), thereby influencing the polarization of the Th phenotype.
(C) VIP
VIP is another neuropeptide with immunosuppressive properties. VIP is one of a group of regulatory molecules termed macrophage-deactivating factors that are believed to prevent the excessive production of pro-inflammatory cytokines (see review by Ganea and Delgado, 2002). VIP inhibits LPS-induced TNF-α, IL-6, and IL-12 production in activated macrophages. It is known that LPS binds to a soluble LPS-binding protein, and this complex then binds to the cell-bound CD14 receptor. VIP induces the rapid shedding of CD14 (Delgado et al., 1999), and this underpins the anti-inflammatory effect of VIP in LPS-driven inflammation such as periodontitis. In addition, VIP stimulates the production of the potent anti-inflammatory cytokine, IL-10, and suppresses T-cell proliferation (Ganea and Delgado, 2002). Non-neuronal sources of VIP have been identified in inflammatory cells such as neutrophils and mast cells (Cutz et al., 1978; O’Dorisio et al., 1980), and it would appear that VIP has an important role in neuroimmunomodulation (Pozo et al., 2000).
(D) NPY
NPY is a potent vasoconstrictor and amplifies the post-synaptic effects of other vasoconstrictors such as noradrenaline (Lundberg et al., 1985). The relationship is complex because NPY inhibits the pre-synaptic release of noradrenaline and vice versa (Zukowska-Grojec and Wahlestedt, 1993). It is believed that NPY acts through at least four receptor subtypes to inhibit sympathetic neurotransmitter release; however, the vasoconstrictor activities of NPY in vivo are mediated principally by the NPY Y1 receptor (Malmström, 2000). In addition, NPY has potent angiogenic properties. At low physiological concentrations, in vitro NPY promotes vessel-sprouting (Zukowska-Grojec et al., 1998) and endothelial cell proliferation (Marion-Audibert et al., 2000), suggesting that it may have a role in tissue development and repair.
(6) Conditions with a Neurogenic Component
The peripheral release of neuropeptides, via antidromic axon-reflexes, is an essential prerequisite for neurogenic inflammation. The concept that sensory nerves may amplify and spread inflammation has been associated with a range of inflammatory diseases and disorders. A role for neurogenic inflammation in asthma has been extensively investigated, and airway sensory nerves have been shown to express several neuropeptides believed to play key roles in this condition. The involvement of neuropeptides in clinically relevant responses such as bronchoconstriction supports a role for the tachykinins in asthma (Lundberg, 1995; Chu et al., 2000). Neuropeptides are also abundant in the alimentary canal, the most important source being the cholinergic neurons of the enteric nervous system. Changes in peptidergic neurons leading to imbalances in neuropeptide expression are believed to be intimately associated with gastrointestinal disease. Alterations in peptidergic innervation of the gut have been associated with gastrointestinal infection (Masson et al., 1996) and inflammation (Kimura et al., 1994). Further evidence supporting a neurogenic involvement in gut inflammation comes from studies showing positive responses to neuropeptide antagonist treatment (Croci et al., 1997). There is also ample evidence to suggest a close association between neuropeptides and inflammatory joint disease. In particular, much research has focused on the role of neuropeptides and rheumatoid arthritis (Kidd et al., 1996). A heterogenous population of sensory nerves is known to innervate synovial joints, with SP and CGRP thought to have particularly important roles (Cruwys et al., 1995) in the neurogenic inflammatory response.
With the extent of current literature on the pro-inflammatory effects of neuropeptides, it is easy to overlook their role in the protective aspects of inflammation, i.e., healing and repair (for review, see Brain, 1997). Healing requires specific organizational and functional reactions of the nervous and microcirculatory systems. Inadequate or inappropriate coordination of these responses is associated with disorders of wound healing. The clinical observation that sensory neuropathies may be associated with persistent skin ulcers (Bockers et al., 1989) lends support to the involvement of neuropeptides in tissue repair. Evidence for a trophic role in dental wound healing comes from studies showing that neuropeptides stimulate the proliferation of gingival fibroblasts (Bartold et al., 1994) and pulpal cells (Bongenhielm et al., 1995). Recently, it has been shown that exogenously applied neuropeptide improved cutaneous wound-healing kinetics in an animal model (Gibran et al., 2002).
(7) Neuropeptides in Oral and Periodontal Health and Disease
The periodontal tissues are innervated by the sensory fibers of the maxillary and mandibular divisions of the trigeminal nerve. Sensory nerve fibers, which make up the great majority of periodontal innervation, originate in the trigeminal ganglion and range in size from C fibers (0.1 to 1.0 μm in diameter) to Aδ (1 to 5 μm in diameter) and Aβ fibers (6 to 10 μm in diameter). At least 10% of the non-myelinated nerves comprise the autonomic supply made up of either sympathetic fibers derived from the superior cervical ganglion or parasympathetic fibers from either the sphenopalatine ganglion for the upper teeth or the otic ganglion for the lower teeth. Periodontal tissues are extensively innervated by myelinated nerve fibers, closely associated with blood vessels. These fibers lose their myelin sheath as they course though the ligament and terminate as free non-myelinated endings or in a number of specialized receptors. The detailed organization of the nerve supply to the periodontal tissues is beyond the scope of this review (see Sinnatamby, 1999).
(A) Neuropeptides in gingival and periodontal tissues
Fibers innervating the periodontal tissues in humans are immunoreactive to a number of neuropeptides, including SP, CGRP, VIP, and NPY (Luthman et al., 1988). SP has been localized by immunohistochemistry in normal human gingival tissues perivascularly and within the rete pegs (Bartold et al., 1994). Nerve fibers originating in the subepithelial connective tissue may also penetrate the junctional epithelium. The intra-epithelial nerves are unmyelinated but contain a number of neuropeptides (Bartold et al., 2000). Previously, the junctional epithelium was shown to be extensively innervated by SP nerve fibers (Nagata et al., 1992). Furthermore, neuroendocrine cells within the epithelial cell rests of Malassez have been shown to express SP, CGRP, and VIP (Kvinnsland et al., 2000).
CGRP, VIP, and NPY have been shown to be associated with blood vessels in the rat periodontal ligament, suggesting a role for these molecules in regulating blood flow in this region (Kato et al., 1996). It has been shown recently that application of capsaicin to oral mucosa induced an axon-reflex-mediated increase in blood flow in human gingivo-mucosal tissues (Kemppainen et al., 2003). The injury-evoked appearance of NPY in myelinated axons in the lingual periodontal ligament of the rat incisor following peripheral nerve injury is suggestive of a participatory role in regeneration (Wakisaka et al., 1996). Increased levels of NPY have previously been reported in the trigeminal ganglion following peripheral nerve injury (Wakisaka et al., 1993). It now appears that the increased NPY in the sensory ganglia may be transported both peripherally and centrally. Further evidence suggests that NPY is also present in other non-sympathetic innervation, associated with the neuropeptide VIP (Norevall and Forsgren, 1999). Thus, following sympathectomy, the effects of sympathetic nerve activity may be mimicked by sensory and NPY/VIP-ergic innervation, highlighting the tremendous plasticity of the peptidergic nervous system.
(B) Neuropeptides and oral disease
Evidence for the development of neurogenic inflammation in the oral cavity was first presented just over a decade ago (Fazekas et al., 1990). Further work showed that SP was a major mediator of the neurogenic inflammatory response in the oral mucosa (Gyorfi et al., 1991, 1992, 1993, 1995). Factors associated with neurogenic inflammation in cutaneous tissues are also present in orofacial tissues. There are important differences, however, between the trigeminal and spinal nervous systems that necessitate the development of specific models to study the potential contributions of neurogenic processes to inflammation affecting oral and dental tissues (Flores et al., 2001). Whereas, during embryogenesis, neurons of the dorsal root ganglia are derived entirely from the neural crest, those of the trigeminal ganglion have origins in the neural crest as well as the neurogenic placodes. Although the physiological significance of this difference is unknown, several studies suggest that there may be substantial differences in the density, morphology, ontogeny, and function of trigeminal compared with spinal nociceptors. For example, ligation or constriction of the sciatic nerve increased sympathetic nerve sprouting in the affected dorsal root ganglia, but similar injuries to the inferior alveolar nerve did not produce similar sprouting in the affected trigeminal ganglion (Bongenhielm et al., 1999). It would appear that there are differences not only in the neurogenic responses between the trigeminal and spinal systems but also between various tissues within the oral cavity (Flores et al., 2001).
(C) Neuropeptides and pulpal disease
Pulpal disease and the potential role for neuropeptides in pulpitis have been extensively reviewed elsewhere (Hildebrand et al., 1995; Byers and Närhi, 1999; Orchardson and Cadden, 2001; Byers et al., 2003). SP, NKA, CGRP, NPY, and VIP have been quantified in healthy human teeth by radioimmunoassay (Awawdeh et al., 2002a; El-Karim et al., 2003). Furthermore, SP, NKA, and CGRP have been shown to be more abundant in pulp tissue from painful compared with healthy human teeth (Awawdeh et al., 2002a), suggesting a possible role in pulpal pain. Neuropeptides may also have a trophic role in the pulp, since innervated teeth with pulp exposures were shown to have much less tissue necrosis and periapical destruction than denervated teeth (Byers and Taylor, 1993).
The interrelationships and correlations between pulpal and periodontal disease have recently been reviewed (Rutherford, 2002). Substantial evidence suggests that pulpal disease can contribute to periodontal disease, but there is more controversy about whether the reverse is true. The pulp connects to the periodontal ligament through the apical foramen, lateral or accessory canals, and possibly through patent dentinal tubules in the presence or absence of cementum. Blood vessels course in lateral or accessory canals connecting the coronal and/or radicular pulp with the periodontal ligament. However, although the pulp and periodontium undoubtedly communicate, many aspects of their pathophysiology remain discrete. Whereas periodontal tissues can regenerate if bacterial plaque is removed, the healing process in the pulp does not recreate the original tissue architecture, perhaps because of the unique low-compliant environment. Thus, heterogeneity between periodontal and pulpal tissues underlines the necessity for the responses of each tissue to be studied separately. Indeed, within periodontal and pulpal tissues, innervation patterns continually change, depending on the maturation or pathophysiological requirements of the microenvironment.
In the context of the current review, it is of interest that morphological and electrophysiologic techniques have shown that some inferior alveolar nerve fibers have branches that innervate both pulp and periodontium (Capra et al., 1984; Foster and Robinson, 1994). Moreover, recent work in our laboratory indicates that increased tachykinin levels in painful human dental pulp are associated with increased tachykinin levels in the GCF of painful and adjacent teeth (Awawdeh et al., 2002b). It is possible that the increased levels of pulpal tachykinins are mirrored by an increase in activity of fibers in the periodontium, leading to higher concentrations of neuropeptides in GCF. Indeed, physiological studies in animals have demonstrated that sensitization of pulpal afferents modulates the sensitivity of periodontal afferents, possibly by means of an axon reflex, which induces neurogenic inflammation in the periodontal tissues (Matsumoto et al., 1996).
(D) Neuropeptides and periodontitis
Periodontitis, a chronic inflammatory condition constituting a major oral health problem, has an important neurogenic component (Gyorfi et al., 1992). The oral cavity is particularly susceptible to external insult, including traumatic challenges, changes in pH, temperature changes, and variable exposure to endotoxin. Persistence of any one of these factors, outside the range of normality, could be enough to stimulate neurogenic inflammation in susceptible tissues. With the recognition that peptidergic neurons extensively innervate the gingivae (Luthman et al., 1988, 1989), and the identification of neuropeptides in GCF (Linden et al., 1997, 2002; Lundy et al., 1999, 2000b; Hanioka et al., 2000), it is becoming increasingly evident that periodontitis and indeed other orofacial inflammatory disorders may be modulated by imbalances in certain neuropeptides. The distribution of neurochemical markers in tissue obtained from gingivitis- and periodontitis-affected sites led Luthman et al. (1989) and Bartold et al. (1994) to suggest a possible role for neuropeptides in the pathogenesis of periodontitis. However, in addition to neuropeptides, the intensity and duration of inflammation in periodontal disease are also controlled by a complex regulatory network of inflammatory mediators and immune cells. As a result, the task of understanding the neurogenic component of periodontal health and disease is all the more challenging, because changes in neuropeptide levels are only part of a superimposed cascade of chemical activity.
In periodontitis-affected subjects, the levels of both SP and NKA were significantly elevated in GCF of disease-affected teeth compared with healthy sites (Linden et al., 1997). In this study, radioimmunoassay was used to quantify SP and NKA in GCF from single sites, avoiding the necessity to pool GCF with subsequent loss of site-specific information. The tachykinins could have biological activity at the levels isolated; however, it remains unclear whether they function in the pocket or in the connective tissue or in both. In this context, the reported immunolocalization of SP specifically to inflammatory foci in human gingiva may be relevant (Bartold et al., 1994). Release of neuropeptides into the gingival connective tissue could be followed by diffusion through the epithelium and subsequently into the gingival crevice or periodontal pocket. Comparable levels of SP were also found in GCF collected from inflamed sites by a competitive ELISA (Hanioka et al., 2000). SP showed a significant correlation with indicators of host response, including PGE2, IL-1β, and TNF-α (Hanioka et al., 2000). SP and NKA are present at very low levels (< 10 pg/mL) and have a very short half-life in serum, and so the source of these neuropeptides in GCF is likely to be from nerve fibers or inflammatory cells present in the local environment. The high levels of SP and NKA in GCF in periodontitis-affected sites have been shown to fall as a result of periodontal treatment (Lundy et al., 2000b), supporting the view that there is a local source of these tachykinins, which is linked to the inflammatory process associated with periodontitis. Plaque may be an important co-factor mediating some of the effects of SP in periodontitis, since it has been shown (Lieb et al., 1996) that SP was able to induce synthesis of IL-6 in monocytes in vitro only when LPS was added. In addition to augmenting cytokine production, SP acts as a pro-inflammatory mediator by limiting the production of TGF-β by LPS-activated macrophages (Marriott and Bost, 1998). We observed considerable inter-individual variability in neuropeptide levels, particularly of SP in periodontitis sites. Inter-individual variability in neuropeptide tissue levels has previously been commented on (Maggi et al., 1987) and may be directly related to the intensity of the efferent function of sensory nerves.
CGRP has also been detected in GCF; however, the levels were increased in periodontally healthy compared with gingivitis sites (Lundy et al., 1999). CGRP was not detectable in any periodontitis site, and it was concluded that components of GCF were responsible for degrading this neuropeptide in diseased sites (Lundy et al., 1999). It was subsequently shown, by matrix-assisted laser desorption time-of-flight mass spectrometry, that carboxypeptidase activity not only initiated but also extensively degraded CGRP (Lundy et al., 2000a). The rapid selective degradation of the anti-inflammatory neuropeptide CGRP, but not SP or NKA (Lundy et al., 2000a), could serve to enhance the pro-inflammatory effects of the tachykinins in periodontal inflammation. It is known that many of the emerging effects of SP on leukocytes are stimulatory, whereas those of CGRP are inhibitory (McGillis and Fernandez, 1999). Carboxypeptidase release by degranulating mast cells would fit the hypothesis, first suggested by Brain and Williams (1989), that SP-evoked release of proteolytic enzymes from local mast cells could lead to accelerated degradation of CGRP. Degradation of CGRP may be particularly relevant to periodontal destruction, since this neuropeptide has been shown to inhibit osteoclastic bone resorption and stimulate osteogenesis (Konttinen et al., 1996).
The levels of VIP reported in GCF support a potential pathophysiological role for this neuropeptide in periodontal health and disease (Linden et al., 2002). VIP was significantly elevated in periodontitis-affected sites compared with the levels in clinically healthy sites. Non-surgical periodontal treatment resulted in a clinical improvement along with a concomitant reduction in the levels of VIP in periodontitis sites (Linden et al., 2002). Since VIP is present at very low levels (< 2 pg/mL) and has a short half-life in serum (Hoyle, 1995), the source of the relatively high levels of VIP in GCF is likely to be nerve fibers or inflammatory cells present in the local environment. It is possible that, during an inflammatory response, the timely release of VIP could act as an autocrine/paracrine factor influencing the down-regulation of the ongoing immune response, whereas SP is known to promote the release of pro-inflammatory cytokines. Opposing roles for SP and VIP have previously been described in a murine model of inflammation in lung parenchyma (Kaltreider et al., 1997).
The role of neuropeptides in exerting a trophic effect on peripheral tissues is also worthy of mention. Such a system has been proposed to act tonically so that normal sensory stimuli would produce a continuous outflow of sensory transmitters whose actions would maintain tissue integrity (Maggi and Meli, 1988). Our studies would support this view, since low levels of SP, NKA, and CGRP were reported in GCF collected from gingival crevices of teeth with a healthy periodontal status (Linden et al., 1997; Lundy et al., 1999), whereas, in keeping with its known trophic effects, higher levels of VIP were detected in healthy GCF (Linden et al., 2002), suggesting that VIP plays an important role in maintaining periodontal health.
(E) Neuropeptides and bone
Disruption of the balance between bone resorption and formation is central to a variety of diseases ranging from rheumatoid arthritis to periodontitis. Systemic hormones such as calcitonin, parathyroid hormone, and vitamin D3 are important in bone metabolism. The local microenvironment is also a significant factor in bone remodeling, and evidence is accumulating that the nervous system may have an important role in this context. The abundant peptidergic innervation of bone may simply reflect sensory function or control of the vascular system; however, it is becoming apparent that neuropeptides may have important modulatory roles in bone metabolism.
Initial interest in neuropeptide involvement in the process of bone metabolism was prompted by the finding that CGRP showed amino acid sequence homology to the N-terminal region of calcitonin. CGRP has been shown to be abundantly distributed in bone via nerve fibers closely associated with blood vessels (Hill and Elde, 1991). In addition, CGRP fibers also show specific regional distribution and have been observed in close contact with osteoclasts (Imai et al., 1997). In the periodontal ligament, CGRP-immunoreactive nerve fibers have been observed extending close to the root cementum (Heyeraas et al., 1993). Moreover, in experimentally induced rat periapical lesions it has been shown that the number of bone-resorbing osteoclasts began to decrease when the density of CGRP-immunoreactive nerve fibers reached their peak (Toriya et al., 1997), suggesting a possible role for CGRP in inhibiting bone resorption. Various experimental studies support the involvement of CGRP in bone remodeling. Hukkanen et al. (1993) demonstrated that CGRP fibers rapidly proliferated in the vicinity of a fracture coinciding with callus formation and subsequent remodeling. CGRP fibers have also been shown to increase in the periodontal ligament in response to mechanical stress induced by orthodontic tooth movement (Kato et al., 1996). Therefore, not only the distribution of CGRP fibers but also their dynamics support an important role for this neuropeptide in bone remodeling (Irie et al., 2002).
The action of CGRP on bone cells has been extensively investigated, and it has been shown to inhibit bone resorption both in vitro (Zaidi et al., 1987) and in ovariectomized rats in vivo (Valentijn et al., 1997). The action of CGRP on osteoclasts appears, at least in part, to involve the inhibition of cell motility (Alam et al., 1991). CGRP modulates the differentiation of osteoblasts and their production of cytokines in vitro (Valentijn et al., 1997). Indeed, histochemical associations between CGRP fibers and osteoblasts have been demonstrated (Imai et al., 1997). Receptors for several neuropeptides, including CGRP, are present on osteoblasts (Bjurholm et al., 1992). In addition, osteoblasts have recently been shown to express CGRP endogenously. Transgenic mice with osteoblasts which overexpress CGRP are characterized by increased bone formation and enhanced bone volume. This suggests that CGRP acts on bone not only via a nervous route but also via an autocrine loop (Imai and Matsusue, 2002). The actions of CGRP on cultured osteoblasts and osteoclasts, together with its in vivo localization, strongly support the paradigm that the nervous system influences bone metabolism.
The presence of neuropeptides in bone tissues has been indicated not only by immunohistochemistry but also by quantification of these neuropeptides in bone by radioimmunoassay (Ahmed et al., 1994). Experimental studies demonstrating that CGRP inhibits osteoclastic bone resorption and stimulates osteogenesis (Konttinen et al., 1996) support a central role for CGRP in bone metabolism. Evidence from our own studies of neuropeptides in GCF revealed an initially surprising lack of CGRP in periodontitis sites (Lundy et al., 1999) as a result of disease-specific carboxypeptidase degradation (Lundy et al., 2000a). In this context, the lack of CGRP could be a pivotal factor for tipping the balance in favor of periodontal bone loss.
(8) Pain and Periodontitis
The periodontal ligament receives a rich sensory nerve supply. Periodontal afferents encode information about both the teeth stimulated and the direction of forces applied to individual teeth. Ruffini endings, which are the primary mechanoreceptors, play a significant role in the specification of the level, duration, and point of attack of forces used to manipulate food between the teeth (Trulsson and Gunne, 1998). Almost a century ago, Sherrington (1906) proposed the existence of the nociceptor, a primary sensory neuron which is activated by stimuli capable of causing tissue damage. The periodontal tissues also contain many nociceptors. Both Aδ and C fibers are present in the periodontal ligament, and their response characteristics suggest that they have a role in periodontal nociception (Mengel et al., 1992, 1993).
Advances have been made in our understanding of the molecular mechanisms through which sensory neurones detect pain-producing stimuli (Julius and Basbaum, 2001). Extracellular protons are the hallmark of the physiological response to injury, and these act on VR1. There are also less-well-characterized nociceptive receptors for other inflammatory molecules, including peptides (bradykinin), lipids (prostaglandins), and neurotrophins (NGF). Little is known about the distribution of nociceptive receptors in the periodontal tissues; however, a recent study has identified the VR1 receptor on about 25% of gingival neurones (Stenholm et al., 2002). Analysis of data from other tissues suggests that VR1 expression is limited to polymodal nociceptors on unmyelinated C fibers but that, during inflammation, Aδ neurones begin to express VR1 (Amaya et al., 2003). VR1 can be considered an integrator of the effects of many noxious stimuli, including heat, extracellular acidification, and vanilloid ligands. It has been hypothesized that there are factors termed endovanilloids, produced under inflammatory conditions, which modulate the response of VR1. Factors such as leukotriene B4, which favor the opening of these ion channels, facilitate pain. Less is known about factors which close VR1 channels and therefore act as endogenous analgesic agents (Di Marzo et al., 2002). It is possible that such ion channel blockers may be involved in the lack of pain associated with periodontal inflammation. Recently, a gene named ‘painless’, which is known to encode a novel member of the TRPV family, has been identified in Drosophila (Tracey et al., 2003). It is not known whether a mammalian homologue exists or whether this gene could have a role in nociceptive signaling in painless conditions such as periodontitis. Further studies are needed to understand how the specific physiological properties of periodontal nociceptors respond to inflammation associated with gingivitis and periodontitis.
Studies are beginning to determine the roles of individual neuropeptides in nociception, and it is now known that SP is required for the full orchestration of the response to pain and injury (DeFelipe et al., 1998). Mice with disruption of the preprotachykinin A gene, which encodes SP/NKA, had significantly reduced responses to moderate or intense pain, and neurogenic inflammation was virtually absent in these mutant mice (Cao et al., 1998). Peripheral administration of SP activates primary afferents, producing nociceptive behaviors, which can be blocked by a SP antagonist (Carlton et al., 1996). An increase in the proportion of axons expressing NK-1 receptors has also been observed during inflammation (Carlton and Coggeshall, 2002). Therefore, it is possible that there could be increased SP-induced activation of peripheral nociceptors in inflamed sites. Despite the increased levels of SP in periodontitis sites (Linden et al., 1997; Lundy et al., 2000b) and the presence of the NK-1 receptor in gingival tissue (Goto et al., 2001), periodontitis is generally painless. It will be important to determine whether natural anatagonists in the gingival microenvironment can block SP binding to the NK-1 receptor. However, the undetectability of CGRP in GCF from periodontitis sites (Lundy et al., 1999) is an important finding in this respect. Recently, it has been shown that the neuropeptide αCGRP is critical for the production and possibly the transmission of somatic and visceral pain signals associated with neurogenic inflammation (Salmon et al., 2001). Mice lacking the αCGRP receptor were shown to display an attenuated response to chemical pain and inflammation (Salmon et al., 2001). This suggests that the absence of CGRP in GCF from periodontally diseased sites (Lundy et al., 1999, 2000b) may partly explain the absence of pain as a major symptom in periodontitis. Furthermore, it is likely that other neuropeptides play important roles in nociception, notably, the requirement for activation of the NPY Y1 receptor to induce peripheral SP release (Naveilhan et al., 2001). Thus, NPY or indeed another Y1 receptor ligand has the potential to mediate antinociception by reducing SP release from primary afferents. It is also important to note that nociceptive signals from the periphery may be amplified or diminished during spinal processing, resulting in either central sensitization or desensitization, respectively. However, until more information is gathered about the potential post-synaptic interaction of neuropeptides and the expression of receptors in periodontal tissue, the mechanisms underlying the painless nature of periodontitis remain to be determined.
(9) Stress and Periodontitis
Although the focus of this review is on peripheral neuropathophysiological mechanisms, functional and structural changes in the CNS may also play a significant role in periodontal health and disease. There is considerable interest in the significance of such central mechanisms and in particular in the role of stress as a modulator of inflammation. Stress generally refers to the condition where coping with actual or perceived stimuli alters the homeostatic state of the organism. The physiological impact of stress is variable across individuals, and susceptibility to stress depends on genetic factors, age, previous stress experiences, and early life events (Shanks and Lightman, 2001). Complex stimuli such as social and physical stressors influence susceptibility to disease by activating a variety of CNS pathways simultaneously. It is not all one-way traffic, since there is a complex network of bi-directional signals linking the nervous, endocrine, and immune systems (Melmed, 2001). The classic response to stress involves activation of the hypothalamic-pituitary-adrenal (HPA) axis, with the net result being increased glucocorticoid production. Overproduction can lead to immune suppression, and it is important to terminate the HPA activation efficiently once environmental demands have been removed (McEwen, 1998). The physiological response to stressors can also modulate the immune system via the autonomic nervous system and by the release of neuropeptides (Breivik et al., 1996). Sustained stress or an inappropriate response, which upsets the normal regulatory homeostatic balance, can alter the susceptibility to disease states associated with immune dysregulation, including inflammatory responses (McEwen, 1998).
In recent years, it has become evident that SP has a significant functional role in the response to stress. Studies of animals with genetic or pharmacological disruption of SP or the NK-1 receptor have focused on more fully defining the functions of SP. NK-1 receptor null mice have an attenuated response to invasion of their territory, suggesting that SP has a role in the adaptive response to stress. Such aggressive responses to territorial challenge have important survival value (DeFelipe et al., 1998). Acute pharmacological blockade, by means of the NK-1 receptor antagonist RP67580, was as effective as the classic anxiolytic agent diazepam in mice (Santarelli et al., 2001). There is the caveat that the majority of studies of stress have been performed in animals; however, the NK-1 receptor antagonist MK869 has been shown to alleviate depression in a placebo-controlled trial in humans (Kramer et al., 1998). Modulation of SP activity could therefore offer a radical new approach to the management of stress, depression, and anxiety.
Experimental stress affects the migration and secretory functions of murine macrophages (Shapira et al., 2000). Stress enhanced the secretion of NO and other inflammatory mediators in response to LPS derived from Porphyromonas gingivalis, thus providing a potential mechanism for accelerated periodontal destruction in stressed subjects (Shapira et al., 2000). It is possible that SP has a role in elevated cytokine production in response to stress. Macrophages harvested from mice subjected to stress show increased spontaneous production of IL-1 and also increased TNF-α and IL-6 production in response to stimulation with LPS. Capsaicin pre-treatment of neo-natal mice diminished such an elevated spontaneous and LPS-driven cytokine production (Chancellor-Freeland et al., 1995; Zhu et al., 1996). The neurotoxin capsaicin eliminates over 90% of sensory afferent unmyelinated C fibers and therefore destroys most of the SP-containing sensory input (Cervero et al., 1984). These experiments suggest that stress may contribute to an inflammatory response through SP-mediating stress-induced cytokine alterations (Chancellor-Freeland et al., 1995; Zhu et al., 1996).
Taken together, the results of these studies support the view that SP is a key effector associated with the response to stress. Peripheral blood concentrations of SP have been shown to increase in response to psychological anxiety in human subjects (Fehder et al., 1997). Hypothetically, stress could also contribute to inflammation through antidromic signaling, resulting in neuropeptide release (Breivik et al., 1996). Thus, potential mechanisms exist, mediated by SP and perhaps other neuropeptides, through which stress could affect the progression of peripheral inflammation and could have involvement in the reported association between stress and periodontitis (see review by Breivik et al., 1996; Linden et al., 1996; Genco et al., 1999).
(10) The Inflammatory Reflex
Since nerve fibers innervate all major organs and tissues, they are well-placed to play a role in regulating the local response to inflammatory stimuli in real time. Compared with the speed of diffusible inflammatory mediators such as cytokines and glucocorticoids (typically hours to days), neural signaling is almost instantaneous (Tracey, 2002). However, nerves may not only have a role in the initiation and sustenance of inflammation but may also play a part in its suppression. Recently, a ‘cholinergic anti-inflammatory pathway’, which inhibits macrophage activation through parasympathetic nerve activity, has been identified. Direct electrical stimulation of the efferent vagus nerve inhibits the synthesis of TNF in major organs (Bernik et al., 2002). The molecular link between the cholinergic nervous system and the innate immunity is a nicotinic, α-bungarotoxin-sensitive (NαBS-R) acetylcholine receptor (Wang et al., 2003). Stimulation of parasympathetic nerves leads to release of acetylcholine, which then interacts with NαBS-R on tissue macrophages, thereby inhibiting the release of TNF, IL-1, and other pro-inflammatory cytokines (Wang et al., 2003). That this receptor is also sensitive to nicotine may shed some light on the paradigm that smokers with periodontal disease generally experience much less acute inflammation (Bergström and Boström, 2001). It is possible that nicotine also dampens the inflammatory response by acting through NαBS-R. The negative effects of smoking on the periodontal tissues, together with the knowledge that smoking actually increases the likelihood of periodontal bone destruction (Grossi et al., 1995; Mullally and Linden, 1996), outweigh any potential benefit from suppressing the local inflammatory response.
(11) Conclusion
The pathophysiology of periodontal disease is complex, and neuropeptides are not solely responsible for the initiation and progression of disease. Multiple signaling systems, which have an element of overlap or redundancy, are required to activate and regulate any neurogenic response. Thus, the degree of inflammation depends not only on the nature of the stimulus and the dose, duration, and route of exposure, but also on the host response, which is subject to various influences, including modulation via neuroendocrine and neuronal pathways. The balance between the various pro- and anti-inflammatory neuropeptides, however, is undoubtedly part of an integrated host response that allows immune and inflammatory cells to communicate and function in the proper spatial and temporal contexts (McGillis and Fernandez, 1999). However, since neuropeptides and inflammatory modulators are released together during the inflammatory response, it will be impossible to assign a specific role to each until work with selective antagonists is completed.
Sensory neuropeptides are ideally suited for an immunomodulatory role because of their wide distribution in peripheral tissues. Neuropeptides are more stable than amine neurotransmitters and therefore have the potential to influence events at local inflammatory sites for longer periods (McGillis and Fernandez, 1999). This fits well with a hypothesis of involvement of neuropeptides as inflammatory mediators influencing effector cells in the localized inflammatory lesions which characterize periodontitis. Further understanding of complex regulatory systems, in particular those governing neuropeptide inactivation, should extend our knowledge of the inflammatory process and provide potentially novel therapeutic approaches to the management of inflammatory disorders such as periodontitis. In particular, enzymes that inactivate neuropeptides are highly relevant targets for the development of inhibitors and could become candidates for drug development.
Factors influencing the release of substance P (SP) from sensory nerves. SP is released from large dense core vesicles (LDCVs) following (1) cleavage of protease-activated receptor-2 (PAR-2) by tryptase, (2) bradykinin binding to B2 receptors, (3) sensitization by cytokines induced by LPS, or (4) nitric oxide. Activation of the vanilloid receptor and sensitization of sensory nerves by prostaglandins are not shown. Exocytosed SP binds to the neurokinin-1 receptor (NK1-R) expressed on endothelial cells causing vaosodilation and edema formation, prominent features of neurogenic inflammation. SP also interacts with immune cells causing T-lymphocyte proliferation. NK-1 receptor expression on macrophages and monocytes is not shown. As a result of these interactions, SP is believed to act as an important mediator of crosstalk between the nervous and immune systems. SP, Substance P; NK1-R, neurokinin 1 receptor; LDCVs, large dense core vesicles (containing neuropeptdies); PAR-2, protease-activated receptor-2; B2, bradykinin receptor; TLR-4, Toll-like receptor-4; LPS, lipopolysaccharide; and LBP, lipopolysaccharide binding protein.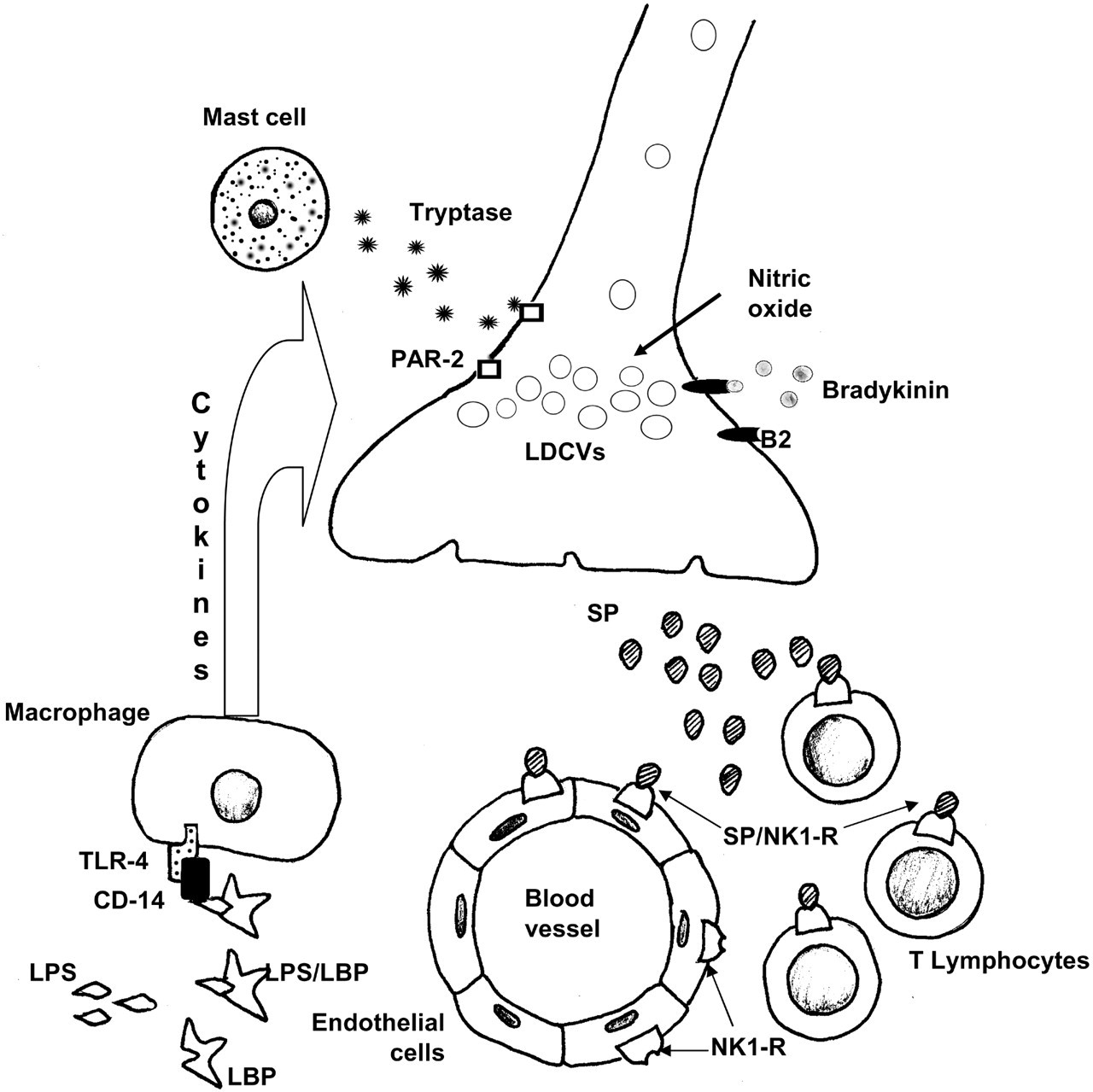
Footnotes
Acknowledgements
This review is based on collaborations with our esteemed colleagues Professor P.-J. Lamey (Queen’s University of Belfast), Professor C. Shaw and Dr. D. Orr (University of Ulster), and post-graduate students, to whom we are greatly indebted.