
Abstract
Proteins found in mineralized tissues act as nature’s crystal engineers, where they play a key role in promoting or inhibiting the growth of minerals such as hydroxyapatite (bones/teeth) and calcium oxalate (kidney stones). Despite their importance in hard-tissue formation and remodeling, and in pathological processes such as stone formation and arterial calcification, there is little known of the protein structure-function relationships that govern hard-tissue engineering. Here we review early studies that have utilized solid-state NMR (ssNMR) techniques to provide in situ secondary-structure determination of statherin and statherin peptides on their biologically relevant hydroxyapatite (HAP) surfaces. In addition to direct structural study, molecular dynamics studies have provided considerable insight into the protein-binding footprint on hydroxyapatite. The molecular insight provided by these studies has also led to the design of biomimetic fusion peptides that utilize nature’s crystal-recognition mechanism to display accessible and dynamic bioactive sequences from the HAP surface. These peptides selectively engage adhesion receptors and direct specific outside-in signaling pathway activation in osteoblast-like cells.
Introduction
Nature has evolved sophisticated strategies for engineering hard tissues through the interaction of proteins and, ultimately, cells with inorganic mineral phases. The remarkable material properties of bones and teeth thus result from the activities of proteins that function at the organic-inorganic interface. The underlying molecular mechanisms that control biomineralization are of significant interest to both medicine and dentistry, since disruption of biomineralization processes can lead to bone and tooth hypomineralization or hypermineralization, artherosclerotic plaque formation, artificial heart valve calcification, kidney and gall stone formation, dental calculus formation, and arthritis, among other pathological calcification processes (Schoen et al., 1988
At the level of fundamental science, it is important to note the paucity of molecular structure information available for biomineralization proteins in general, and in particular for mammalian proteins that directly control calcification processes in hard tissue. Even the most fundamental questions about how the proteins interact at the biomineral surface—such as their general structure and orientation on the calcium phosphate surfaces, or whether the acidic residues are truly interacting directly with the crystal surface—remain largely uncharacterized at the experimental level. To develop a better structure-function-level understanding of protein-crystal molecular recognition, we have begun to utilize solid-state NMR techniques to determine the molecular structure of proteins and peptides on calcium phosphate surfaces. These same techniques have provided interesting molecular dynamics information for the proteins on the biomineral surface. In this review, we will highlight recent work that is providing insight into the structure and crystal recognition mechanisms of the salivary protein model system, but which also provides a general approach to the study of protein-crystal interactions in molecular detail. In addition, the molecular insight into nature’s strategy for crystal recognition has led to the design of peptides that connect the basic studies to applications in the biomaterials arena.
Statherin Background
We focus in this review on a salivary protein that has evolved to control the nucleation and growth dynamics of hydroxyapatite in saliva. Statherin inhibits both the nucleation and the growth of hydroxyapatite in the supersaturated environment of saliva (Schlesinger and Hay, 1977; Hay et al., 1984; Campbell et al., 1989; Johnsson et al., 1991; Jensen et al., 1992; Raj et al., 1992a,b; Schwartz et al., 1992). The statherins and histatins have historically served as two of the best-characterized models for understanding the functional activities of proteins in the physical-chemical control of hydroxyapatite growth. The N-terminus of statherin contains the DSpSpEE (where Sp is phosphorylated serine) acidic motif that is also found in larger proteins such as osteopontin that contain both HAP-binding domains and integrin-binding domains, as well as in non-mammalian biomineralization proteins that function in very different mineral settings (Waite and Qin, 2001).
The effects of native statherin on hydroxyapatite growth dynamics have been extensively characterized by Nancollas and others (Aoba and Moreno, 1984; Johnsson et al., 1991). Nancollas has demonstrated that N-terminal peptides from statherin also display functional activities in controlling HAP growth (Raj et al., 1992a,b; Wikiel et al., 1994). The statherins also serve an important functional role as boundary lubricants (Douglas et al., 1991) and in periodonto-pathology as mediators of bacterial adhesion (Amano et al., 1996; Nagata et al., 1997). Secondary sequence predictions suggest that the N-terminus has a propensity for α-helix formation, and circular dichroism studies have demonstrated the presence of some α-helical conformation in solution (Douglas et al., 1991; Gururaja and Levine, 1996). Statherin has been reported as unstructured in aqueous solution by solution NMR techniques (Naganagowda et al., 1998), although the N-terminal 15-amino-acid domain was found to have some α-helical structure in the structure-inducing solvent trifluoroethanol.
Structure of Statherin on HAP
Some of the key questions driving our studies involve the inter-related aspects of protein conformation on and off the HAP surface, the binding “footprint”—or which amino acid side-chains actually come into contact with the surface, the role of water at the protein-crystal interface, the dynamics of proteins on the mineral surface, the orientation of the protein on the crystal surface, and finally the question of how structure and orientation are related to function. The former question of how protein conformation changes on the crystal surface is connected to an important challenge faced by acidic proteins that control the nucleation and/or growth of biominerals. On the one hand, the structurally organized display of carboxylate side-chains could potentially be used to match the protein electrostatic surface with the complementary ionic lattice of the biomineral, leading to “capping” of growth sites. At the same time, such an organized display of ion-binding side-chains could also lead to binding stabilization of early crystal nucleation clusters and promotion of crystal growth. Such a dual activity for salivary proteins has indeed been demonstrated by Campbell and Nancollas (Campbell et al., 1989).
A potential route to solving this challenge is for the protein to be relatively unfolded in solution, and to utilize crystal-binding energy to stabilize a folded conformation where the side-chains are optimized for interactions with the crystal surface (Fig. 1). We have begun to test whether statherin matches this structural profile by determining its structure on model HAP crystals. Three complementary structural techniques were utilized for determination of the backbone structure of the N-terminal binding domain of statherin. Dipolar Recoupling in a Windowless Sequence (DRAWS) and Double Quantum DRAWS (DQDRAWS) are homonuclear dipolar recoupling techniques that measure the distance between adjacent backbone carbonyl carbons. These distances provide a model-free determination of the backbone torsion angle φ, which can be directly related to local secondary structure and also to the structural heterogeneity at specific backbone atoms. These techniques are particularly powerful when combined with Rotational Echo Double Resonance (REDOR), a heteronuclear recoupling technique that provides distance measurements across putative α-helical or β-sheet hydrogen bonding interactions (i.e., i to i+4 positions in a helix). It should be noted that while isotropic chemical shifts of proteins can yield qualitative indications of secondary structure in homogeneous systems, their values can be influenced by the bulk magnetic susceptibility of inorganic surfaces. We have found that chemical shift analysis is thus of limited value in determining secondary structures for peptides and proteins on HAP. Fortunately, the dipolar recoupling techniques provide an independent and more quantitative approach that has yielded the desired structural information.
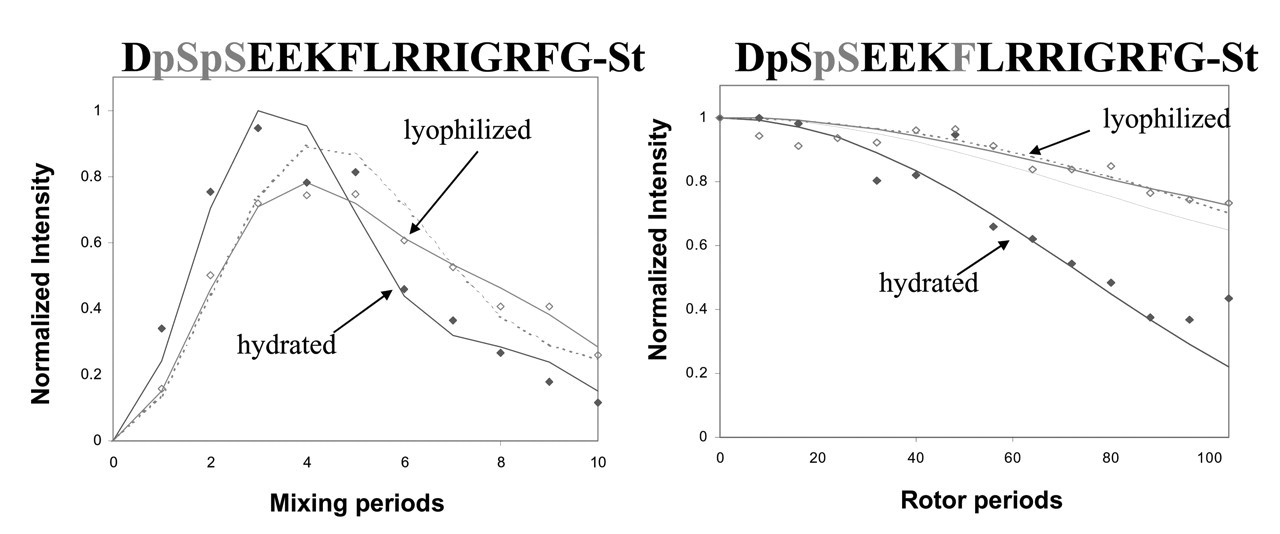
So that the molecular backbone structure of statherin on HAP could be probed under buffered, hydrated conditions, isotopic labels were incorporated at the pS2, pS3, F7, L8, I11, and G12 backbone carbonyl carbons or nitrogens of statherin (Fig. 1). The backbone φ angle was determined with the DRAWS and DQDRAWS techniques at the pS2pS3 positions, while REDOR was used to measure the (i) to (i+4) distance with pS3F7 and L8G12 labeling schemes (Fig. 2). The composite results clearly define this N-terminal, 12-amino-acid binding domain as α-helical on the HAP surface. The strongly acidic N-6 region displays a nearly ideal α-helical distance of 4.2 Å across the pS3F7 hydrogen bond, while the helix is more extended at 4.8 Å across the L8G12 hydrogen bonding position. Low structural dispersion was observed, particularly at the acidic N-terminus, but also out to the 12th amino acid, which suggests that there is a relatively narrow range of molecular structures on the surface.
The same samples were lyophilized under low vacuum (100 mT) after the initial structural studies were completed. A significant lengthening of the pS3F7 distance was observed, demonstrating loss of α-helical structure in this highly acidic region (Fig. 2). The L8G12 distance remained the same, however. These results point to the importance of water in either mediating the interaction of the acidic side-chains with the HAP surface (vide infra), or in directly stabilizing the α-helix conformation (or both). Similar results were observed in recent solid-state NMR studies of N-terminal statherin peptide fragments that were conducted in the lyophilized state both on and off the HAP surface (Long et al., 1998; Shaw et al., 2000a,b). The acidic N-terminus of the peptide was in an extended conformation both on and off the surface, while residues 7–12 were in a partly helical conformation.
It has been previously proposed that the α-helix motif could be used as a scaffolding mechanism for aligning acidic side-chain residues with HAP (Hauschka and Carr, 1982; DeOliviera and Laursen, 1997) by a lattice matching mechanism, or it could align with the surface through a more general electrostatic complementarity. The initial structural characterization shows that the statherin N-terminal binding domain is indeed helical, and further demonstrates the importance of water in the acidic N-terminus interaction with the HAP surface. While the comparison with the solution structure of statherin must be made with caution, because the solution structure is not yet available to the same precision, it appears that the N-terminus is more structured on the surface at the phosphorylated serines and carboxylate-containing aspartic and glutamic acid positions. If verified, this differential folding at the acidic domain between the unbound and bound states would be consistent with a functional role for structural disorder in crystal engineering by acidic proteins, perhaps in response to the aforementioned challenge of matching side-chain positions to inhibit crystal growth without promoting nucleation.
Molecular Dynamic Properties of Statherin on HAP
In addition to structural characterization, the solid-state NMR studies provide interesting molecular dynamics information. This information is particularly useful because the dynamic properties at different atomic positions can be obtained for the comparison of different regions of the N-terminal domain of statherin. As a result, the dynamic studies have directly provided information on the statherin “binding footprint”, or which parts of the protein are in close binding contact with the HAP crystal surface. Initial dynamics studies were conducted on hydrated N15 statherin peptides on HAP crystals. The 13C isotopic backbone labels are sensitive to dynamic timescales ranging over several orders of magnitude, and complementary dynamic characterization techniques include measurement of chemical shift anisotropies (CSAs), T 1 ρ relaxation constants and cross-polarization efficiencies. The principal elements of the motionally pre-averaged CSA tensor provide dynamic information on time scales fast compared with the CSA (i.e., >>20 kHz at a magnetic field of 11.75 Tesla). The T 1 ρ measurements extend the dynamic information available to faster time scales falling between 10−3 and 10−5 seconds.
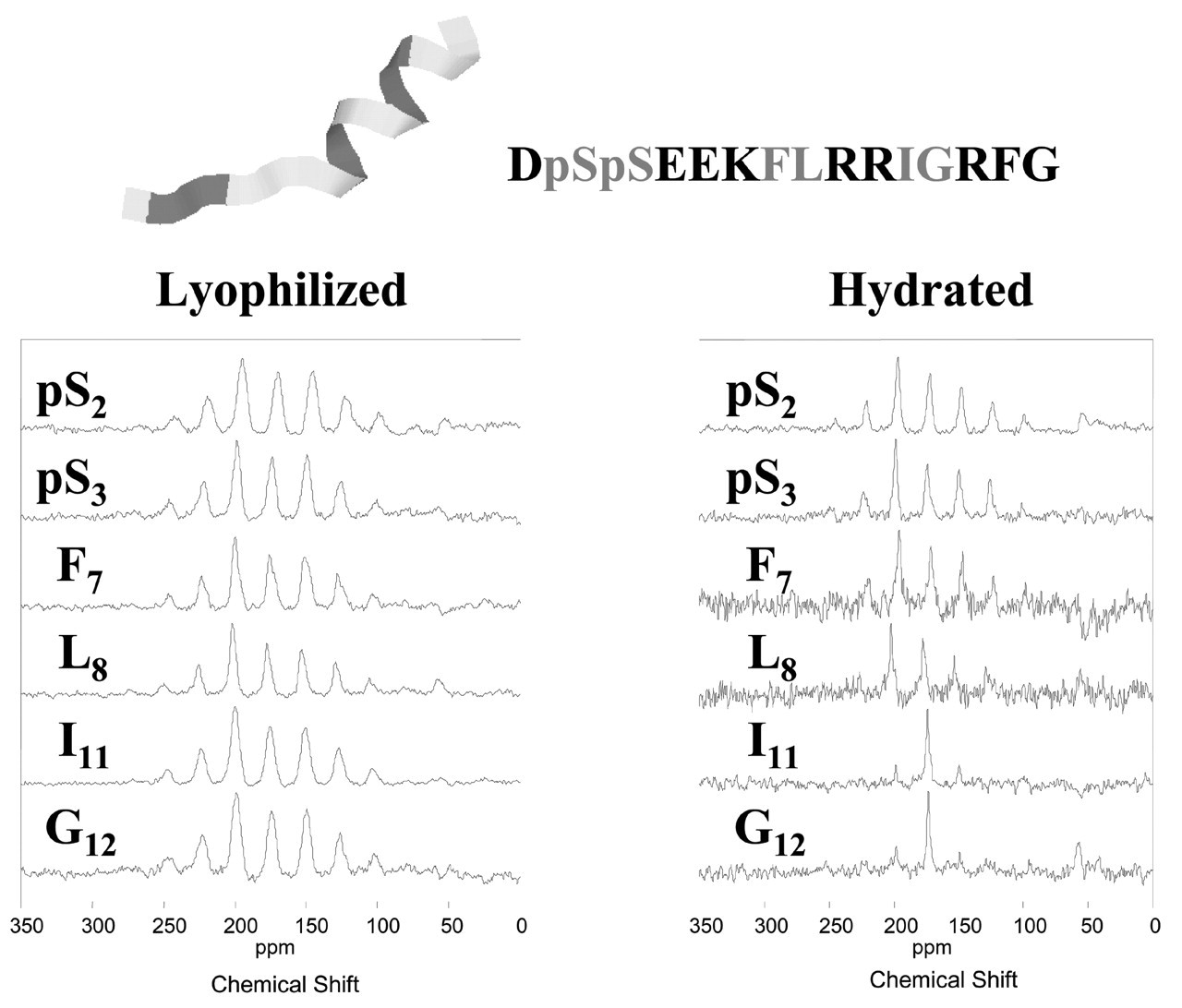
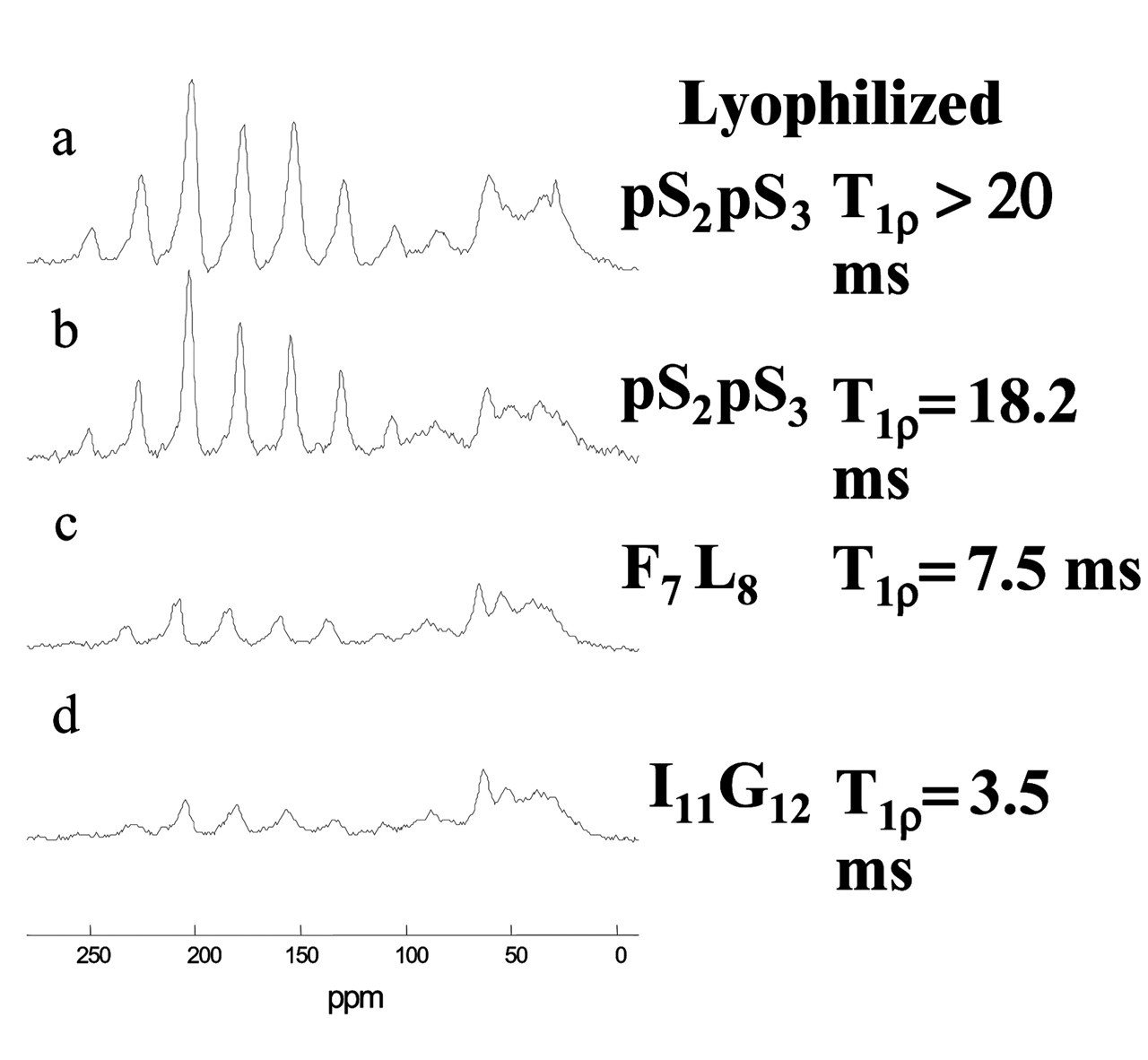
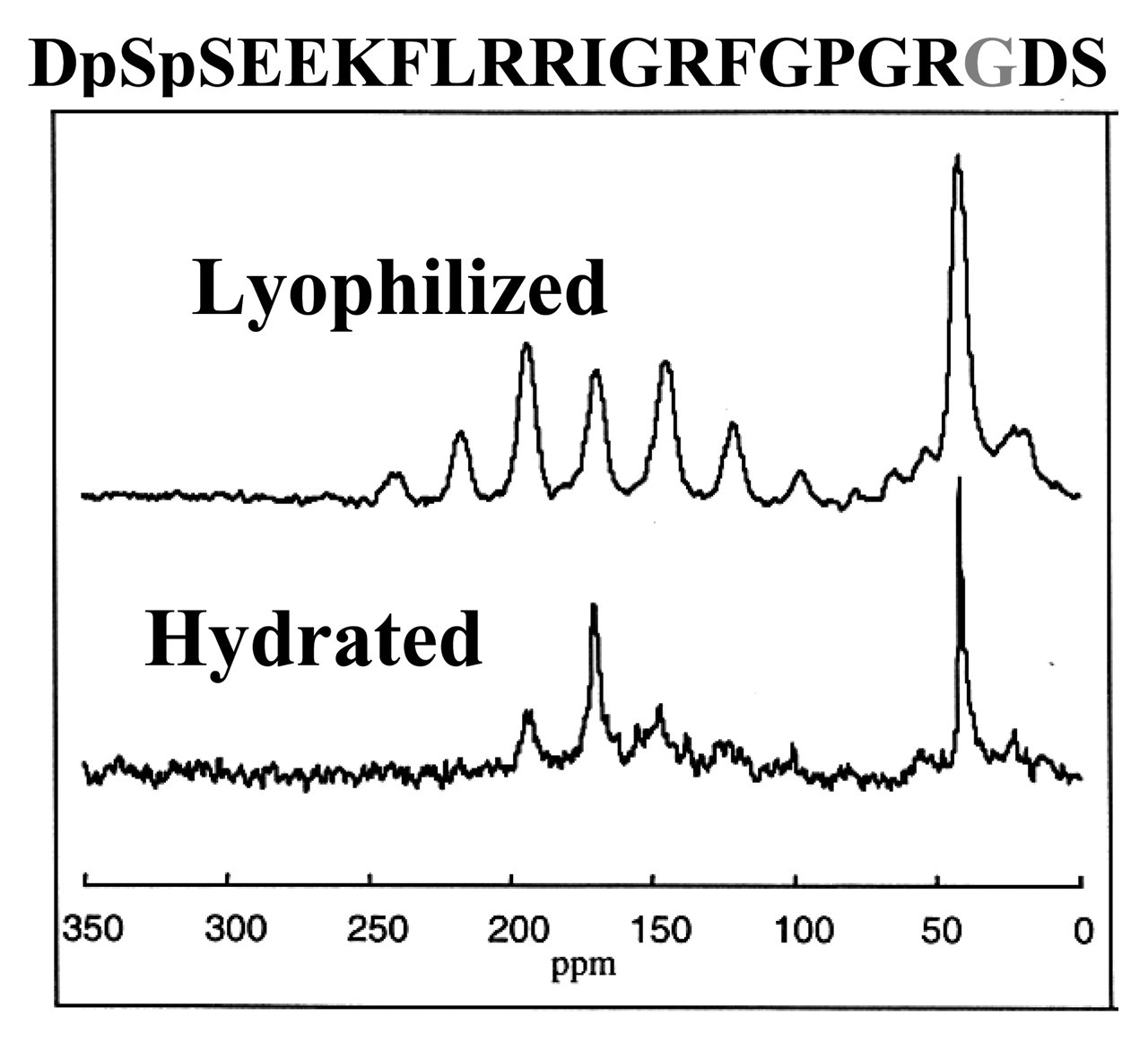
The N15 statherin peptide was characterized at six labeled positions ranging from the N-terminus to near the C-terminus. Fig. 3 compares the spectra obtained for lyophilized vs. hydrated samples at these six positions. As expected, there is a large overall increase in peptide dynamics in the hydrated samples, except at the phosphoserine backbone positions, which remain strikingly immobile in both states. There is increasing motion as the label is moved toward the C-terminus, with large-amplitude dynamic frequencies measured on the order of or greater than 10−5 seconds at the I11 and G12 positions. 13C T 1 ρ measurements provided complementary information, with values for the lyophilized surface-adsorbed samples of > 25 msec, demonstrating that there was little motion on the kilohertz timescale. Similar values of > 25 msec were obtained for the hydrated pS2pS3 positions, while significantly shorter values of 11 msec and 3 msec were measured for the F7L8 and I11G12 positions, respectively. These results at the middle and C-terminal ends of the peptide demonstrated increased dynamic frequencies of greater than 103 Hz. The peptide is thus strongly bound at the acidic N-terminus, but is surprisingly mobile and dynamic at the middle and C-terminal regions.
More recently, similar studies have been conducted at these same positions within the context of the full-length statherin on HAP crystals (Long et al., 2001). The N-terminus is again strongly bound to the crystal surface at the phosphoserine positions, since 13C T 1 ρ values were similar for the lyophilized and hydrated states, with similar cross-polarization efficiencies (Fig. 4). In the middle and C-terminal regions of this statherin domain, there is a significant reduction in T 1 ρ relaxation times, along with a significant loss of cross-polarization efficiencies at the F7, L8, I11, and G12 positions in the hydrated samples. Taken together, these studies demonstrate that the regions outside the acidic N-terminus display protein dynamic modes with frequencies of 10−3 to 10−5 sec, similar to those measured for the N15 peptide. While these results are qualitatively similar to those achieved with the N15 peptide, a comparison of the CSA at the I11 and G12 positions shows that the statherin dynamic mode exhibits a smaller amplitude, since it is nearly unchanged compared with the significantly narrowed CSA for the peptide. The smaller amplitude is consistent with the larger size of the full protein, but could also include differences due to the stabillization of the backbone dynamics by tertiary folding interactions, protein-protein interactions on the crystal surface, or different side-chain interactions with the crystal surface. It is clear from these dynamic studies that the binding footprint is largely confined to the anionic stretch of phosphorylated serines and acidic side-chains at the N-terminus.
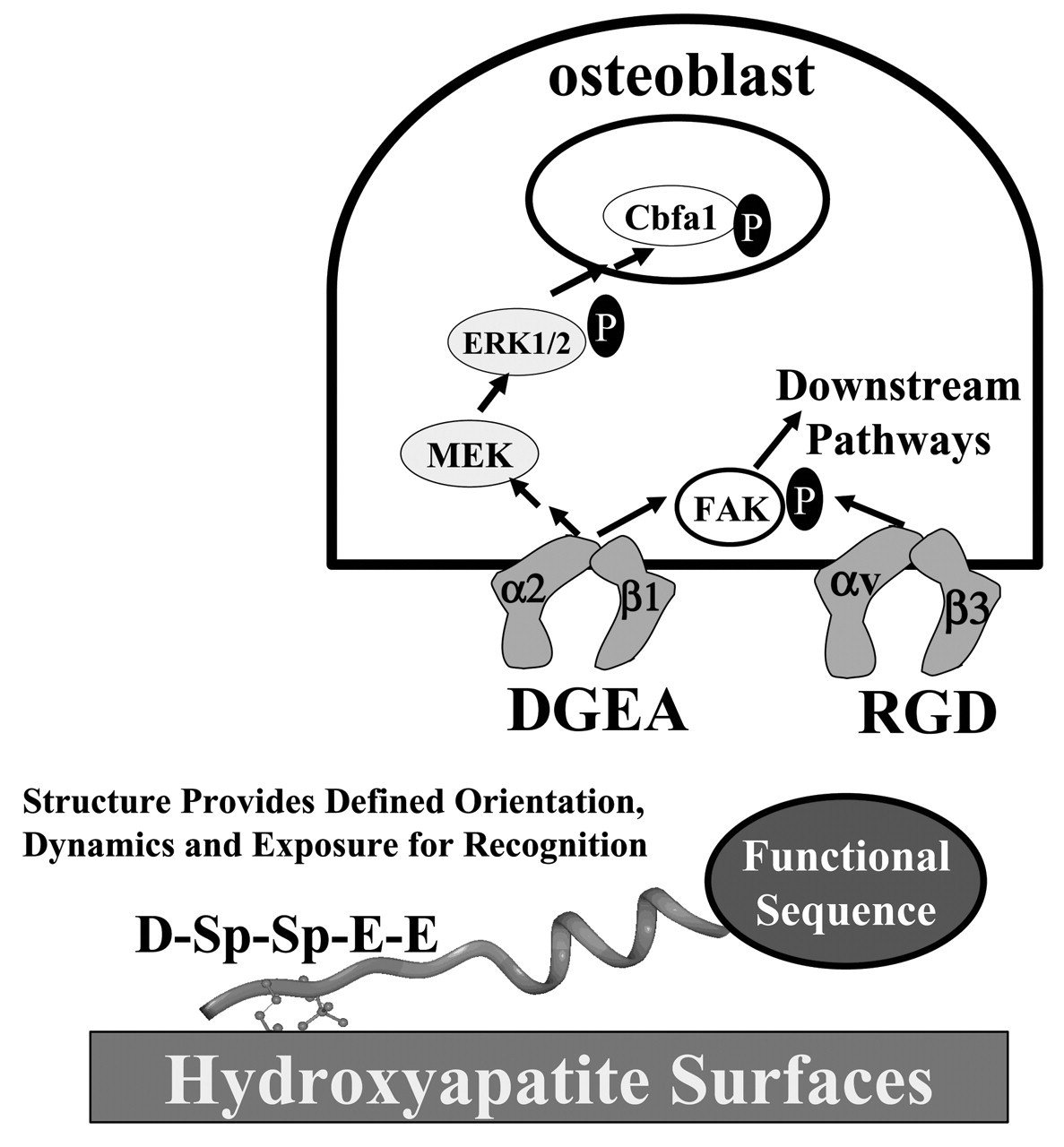
Chimeric Statherin/Biofunctional Peptides for Biomaterial/Tissue Engineering Coatings
The insights gained from the structural and dynamic studies of N15 peptides and full-length statherin have led us to the design of new bioactive peptide coatings for biomaterials and tissue engineering scaffolds (Gilbert et al., 2000). These designs were aimed at taking advantage of the defined α-helical structure toward the C-terminus of the N15 domain, coupled with its mobility and weak interactions with the crystal surface (Fig. 5). The general design couples the HAP-binding sequences from statherin with the addition of bioactive peptide sequences that are displayed with favorable orientation and dynamics. Our initial example is the display of integrin-binding sequences to direct osteoblast behavior (e.g., differentiation and proliferation) on peptide coatings on biomaterials or tissue-engineering scaffolds. In hard tissues, several important extracellular matrix proteins have the interesting property of both recognizing the inorganic phase and displaying domains that engage target cell receptors. Proteins such as osteopontin, bone sialoprotein, and osteocalcin recognize HAP through highly acidic domains, while also displaying adhesive domains that direct engagement of cell adhesion receptors (or, as with statherin, bind other proteins that direct bacterial adhesion). The N15 fusion peptides are designed to mimic these dual functions in a minimized sequence format.
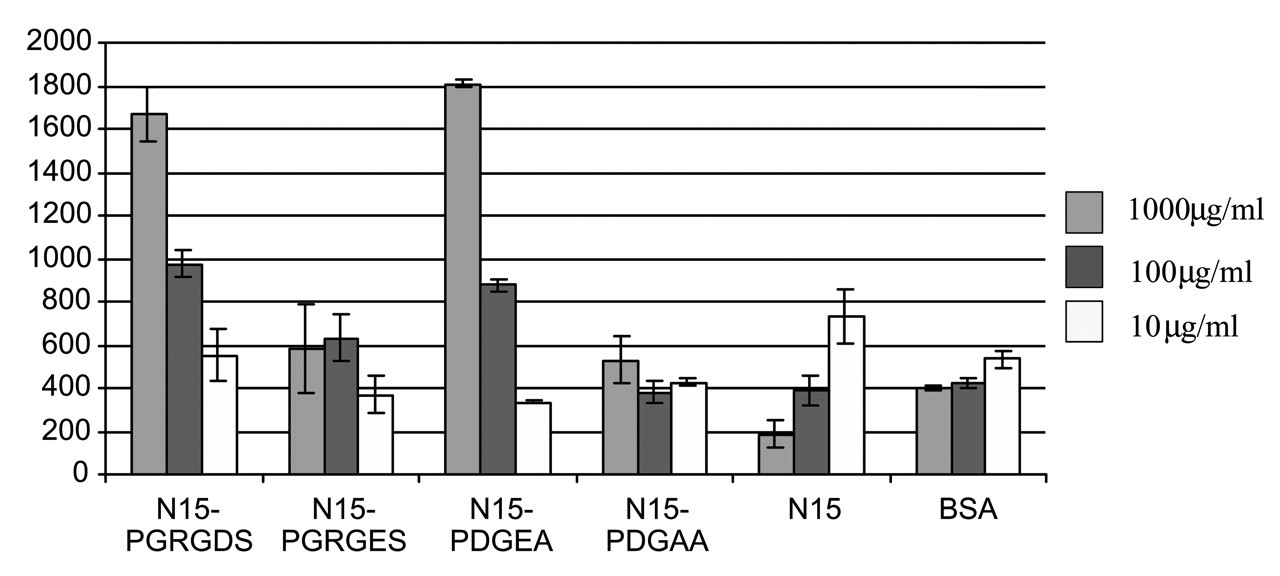
The N15 domain recognizes hydroxyapatite through the N-terminal acidic pentapeptide sequence that contains two phosphorylated serines. This acidic region also provides electrostatic complementarity to drive assembly on cationic polyelectrolyte surface coatings such as polylysine. The arginine-glycine-aspartic acid (RGD) sequence and flanking residues from osteopontin were first added to the N15 C-terminus. The fusion peptide bound as tightly to HAP as the isolated N15 peptide, and directed the RGD-dependent and αvβ3-mediated adhesion of a model integrin-sorted Moαv melanoma cell line. Solid-state NMR experiments showed that the mobility and dynamics of the RGD portion of the hydrated fusion peptide are retained on the HAP surface, despite the presence of the acidic aspartic acid side-chain (Fig. 6). These dynamic properties are likely key to the excellent functional activities of the fusion peptides.
The activity of this RGD-peptide next led us to ask whether peptides could be designed to direct anchorage-specific signaling pathway engagement in an MC3T3-E1 osteoblast-like cell line (Gilbert et al., 2003). Because of the important role of collagen I in the bone matrix, the SN15-PDGEA peptide was selected for comparison with the SN15-PGRGDS peptide (Fig. 7). Antibody and cell adhesion assays demonstrated that the MC3T3-E1 cells were preferentially bound to the SN15-PGRGDS peptide with the αvβ3 integrin rather than the α5β1 fibronectin integrin, and that the α2β1 integrin was the primary target of the SN15-PDGEA peptide. While both the SN15-PGRGDS and SN15-PDGEA peptides directed adhesion events leading to FAK phosphorylation, only the SN15-PDGEA peptide led to MAPK pathway activation as monitored by ERK phosphorylation. These peptides might thus find applications as tissue-engineering or biomaterial coatings that direct integrin-specific signaling pathway activation.
Summary
A molecular model consistent with the current structural and dynamic studies is shown in Fig. 8. The N-terminal domain from residues 2–12 has been shown to be helical on the surface. The strong interaction of statherin with HAP is mediated by the acidic N-terminus, where two phosphoserines and three carboxylate-containing side-chains are located. The dynamics studies suggest that the remainder of the helix is much more weakly interacting with HAP. The mobility of the statherin on HAP could underlie functional observations made previously by others in the field. Moreno and co-workers (1979) found that statherin inhibited secondary crystal growth at surface coverages lower than expected based on the theoretical surface area of the fixed, globular protein. The mobility of statherin on the surface could allow it to block more nucleation sites more effectively than a very rigidly bound protein. The dynamic nature of statherin on the surface may also play a significant role in the lubricating properties of the protein (Ramasubbu et al., 1993). For adequate modeling of the observed motions at the molecular level, further details of the surface-bound tertiary structure and experimental elucidation of the side-chain interactions with HAP will be required to determine the orientation of statherin at the surface. Solid-state NMR does provide a direct route to this information, and we are currently probing the tertiary structure and the interactions of side-chains with the surface for further elucidation of binding mechanisms.
Strategies for determining statherin structure on HAP and summary results for the N-terminal domain. The sequence of statherin is given, along with the 13C-labeled amino acid positions. DRAWS and DQDRAWS determine the backbone torsional angle phi at the pSpS position, while REDOR was used to determine the (i) to (i+4) 13C-15N hydrogen bonding distances between S3 and F7, and L8 and G12. Peptides were synthesized by standard Fmoc solid-phase peptide synthesis strategies, and phosphorylation was performed after protein synthesis for serine 13C=O enriched samples. The HAP crystals had a surface area of 80 m2/g, and adsorption of statherin was carried out at 40 μM for two hours, followed by extensive washing in PBS to remove loosely adsorbed protein. DQDRAWS and REDOR characterization of statherin structure on HAP. The left panel displays the results of the DQDRAWS experiment to measure the backbone angle phi at the pS-pS position. The filled diamonds are data on the hydrated sample, the open diamonds are for the same sample that was subsequently lyophilized, and the dotted line is a simulated data fit to 45% α-helix and 55% β-sheet. The data were taken at room temperature on a 500-MHz home-built spectrometer with a 4-kHz spinning speed, a 34-kHz 13C RF field, and 100-kHz proton decoupling. The REDOR experiment shown in the right panel was performed on a Chemagnetics Infinity 300 spectrometer at a spinning speed of 4 kHz, a 43-kHz 13C RF field, a 45-kHz 15N RF field, XY8 phase cycling on both channels, and 70-kHz decoupling. Measurements on the hydrated samples were done at −25°C to remove any motions on the timescale of the REDOR experiment (as verified by T
1
ρ relaxation measurements). The solid lines represent fits to distances of 4.2 Å for the hydrated sample and 5.2 Å for the sample that was lyophilized after adsorption in buffered solution. The dotted lines are fits to 5.2 Å and 45% α-helix, 55% β-sheet. Dynamic studies of the statherin N15 peptide on HAP. 13C CPMAS spectra of N15 peptides that were adsorbed to HAP crystals of 77 m2/g surface area from 2-mM peptide solutions in modified PBS (100 mM NaCl, 40 mM KCl, 4.3 mM Na2HPO4, and 1.4 mM KH2PO4). After the hydrated sample data were collected, the same samples were frozen and lyophilized in the rotor. Each spectrum consists of 10,000 scans taken on a spectrometer operating at a 125.74-MHz 13C frequency with a 3-kHz sample spinning rate. Dynamic studies of statherin on HAP crystals. CPMAS spectra were acquired at room temperature on a home-built 500-MHz spectrometer with spinning speeds of 3, 4, and 5 kHz. Cross-polarization was accomplished with a 2-msec contact time at RF fields of 50 kHz, and data were collected during 100-kHz proton decoupling. T
1
ρ measurements were done at a spinning speed of 6 kHz on a Chemagnetics Infinity 300 with 13C spin lock times of 0.05 to 4.55 msec at an RF field of 42 kHz after 1.5 msec of cross-polarization with 70-kHz proton decoupling during acquisition. Schematic design of N15 fusion peptides to display integrin-binding sequences. The functional DGEA and RGD sequences from collagen 1 and osteopontin, respectively, were fused to N15 with a proline linker. Subsequent signal pathway studies have shown that only the DGEA fusion peptide activates ERK1/2 phosphorylation, while both peptides direct FAK phosphorylation. Solid-state NMR characterization of the RGD domain dynamics of the N15 fusion peptide on HAP. 13C CPMAS spectra were collected on a Chemagnetics CMX Infinity spectrometer operating at a 13C frequency of 125.72 MHz using a Chemagnetics doubly-resonant MAS probe. These experiments used a 1H 90° pulsewidth of 7.5 msec followed by a contact time of 1.5 msec and a spinning speed of 3003 Hz. The surface-adsorbed samples were signal-averaged for 10,240 scans. The C-terminal glycine retained the large-amplitude dynamics typical of the N15 C-terminus, suggesting that the aspartic acid is not strongly interacting with the HAP surface. The RGD domain dynamics at this position may underlie the excellent cell-binding activity of this peptide when immobilized on HAP surfaces. Characterization of MC3T3-E1 cell adhesion to HAP-immobilized N15-fusion peptides. Both the N15-DGEA and N15-RGD peptides bind the MC3T3-E1 cells in a dose-dependent fashion, while control N15-DGAA and N15-RGE peptides do not bind cells above background control levels. Antibody-blocking studies demonstrated that immobilized N15-PGRGDS bound MC3T3-E1 osteoblasts predominantly via the αvβ3 integrin, and immobilized N15-PDGEA bound MC3T3 E1 osteoblasts predominantly through the α2β1 integrin. Molecular model of the statherin N-terminal domain. The positions used in the REDOR characterization of Fig. 2 are colored. The first red residue is Ser 3, and its corresponding REDOR partner label is Phe 7, colored blue. The second distance was measured from the red position at Leu 8 to the blue position at Gly 12.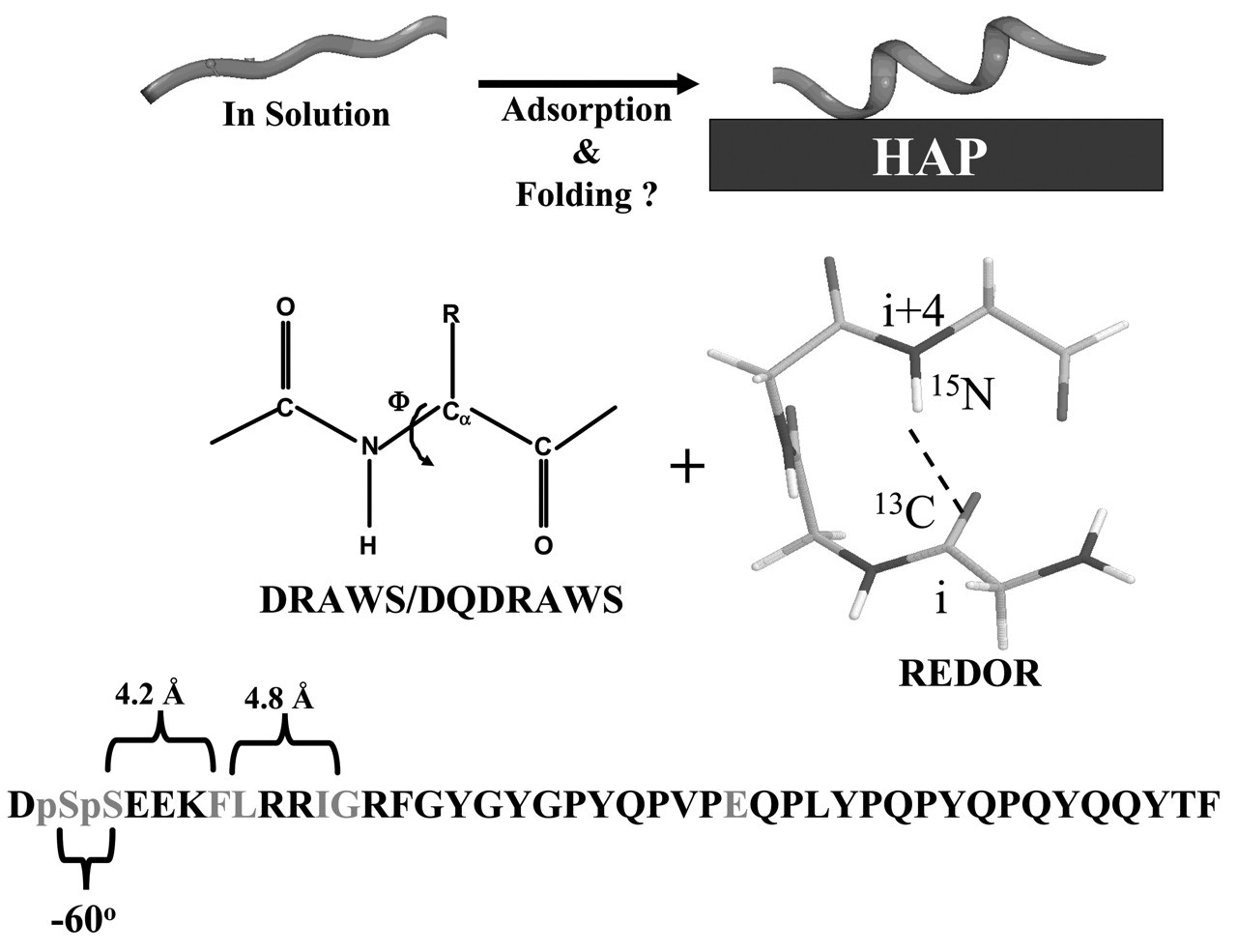

Footnotes
Acknowledgements
We gratefully acknowledge the support provided by the National Institute of Dental and Craniofacial Research (DE 12554), the National Science Foundation (DMR-9616212 and EEC-9529161), and The Whitaker Foundation (graduate fellowship to M.G.). Some of the NMR experiments were performed at the Environmental Molecular Sciences Laboratory (a national scientific user facility sponsored by the DOE Office of Biological and Environmental Research), located at the Pacific Northwest National Laboratory, operated by Battelle for the DOE.