
Abstract
Advances in bacterial genetics came with the discovery of the genetic code, followed by the development of recombinant DNA technologies. Now the field is undergoing a new revolution because of investigators’ ability to sequence and assemble complete bacterial genomes. Over 200 genome projects have been completed or are in progress, and the oral microbiology research community has benefited through projects for oral bacteria and their non-oral-pathogen relatives. This review describes features of several oral bacterial genomes, and emphasizes the themes of species relationships, comparative genomics, and lateral gene transfer. Genomics is having a broad impact on basic research in microbial pathogenesis, and will lead to new approaches in clinical research and therapeutics. The oral microbiota is a unique community especially suited for new challenges to sequence the metagenomes of microbial consortia, and the genomes of uncultivable bacteria.
Introduction
The reductionist approach, the paradigm for bacterial genetics, is exemplified by the study of individual genes and their products. Ingenious and elegant experiments with bacterial mutants led to discoveries of biochemical properties, gene-protein interactions, metabolic pathways, and models of gene regulation that were subsequently validated through recombinant DNA technology and molecular biology. With the advent of whole genome sequencing, the next frontier, the holistic approach of determining how all the genes of an organism interact, has become a possibility. The concept of whole genome sequencing was initiated with the Human Genome Project, and model bacterial genomes were chosen for proof of principle feasibility studies of random shotgun DNA sequencing and assembly strategies. The rest is history, as they say, and the number of microbial genome sequencing projects has grown exponentially since 1995, when the genome sequence of Haemophilus influenzae was published (Fleischmann et al., 1995), to approximately 63 finished and 176 ongoing projects listed in The Institute for Genomic Research (TIGR) Microbial Database (as of May 20, 2002), representing more than half a million genes. On this list of medically and environmentally important micro-organisms are at least 10 oral bacteria spanning the spectrum from known and putative pathogens, e.g., Streptococcus mutans and Porphyromonas gingivalis, to so-called beneficial organisms such as S. sanguis.
Why Sequence?
The genome sequence is the blueprint of all the working parts of a bacterium, and its completion is the first step toward determining how all the parts function and interact. DNA sequence reveals not only the open reading frame (ORF) encoded within a gene, but also associated promoter elements, and thus information necessary to study how expression of a gene is regulated. For virulence genes, both promoter and structural sequences are potential targets for new drugs and vaccines. An increasingly important and practically useful consequence of the growing number of genome sequences is our ability to compare sequences and functions across a wide spectrum of bacteria. With comparative genomics, it is possible to: track the spread of transposons, antibiotic resistance genes, and extrachromosomal elements between species; and analyze the evolution of both metabolic pathways and virulence. Such investigations have demonstrated the large role played by lateral (horizontal) gene transfer in bacterial evolution, and brought to prominence the larger question of what constitutes a species if the genome is a chimera comprised of clusters of genes transferred from unrelated bacteria. On a more practical level, genome sequence information will yield better probes for the detection of pathogens, and eventually permit identification of specific pathogenic strains.
The Process
Microbial genomes are now sequenced exclusively by the bottom-up, random shot-gun strategy first used to derive the sequence H. influenzae (Fleischmann et al., 1995). Typically, a genome project consists of the four phases shown in Fig. 1. First, a genomic library is prepared of small fragments (ca. 2 kb) obtained by mechanical shearing of chromosomal DNA isolated from the organism of interest. Mechanical fragmentation minimizes the biases often observed in libraries made from restriction-enzyme-digested DNA, the result of some enzyme sites being more susceptible than others to digestion. The small DNA fragments are ligated to a high-copy-number bacterial plasmid, usually a pUC vector, and transformed into Escherichia coli. Concurrently, larger insert libraries are prepared in bacteriophage lambda or pBR322-based vectors, and used later in the genome assembly phase as back-up sequencing templates to close any sequence or physical gaps. In the second phase, the ends of all the small fragments are sequenced from vector-specific sequencing primers with the goal of achieving approximately eight-fold coverage of the genome with small random sequences. For example, the P. gingivalis genome is 2.4 Mb; therefore, approximately 35,000 sequencing runs with an average of 500 readable bases per run were required for almost eight-fold coverage. In the third phase, the random sequences are assembled, usually beginning with 30-bp overlaps, until increasingly larger fragments are built and ultimately joined to yield one contiguous sequence. Open reading frames comprised of at least 100 amino acids are identified and assigned a function, first by automated annotation based on the Basic Local Alignment Search Tool algorithm for protein similarity (BLASTP; Altschul et al., 1997) and followed by human visual inspection for verification and decisions on questionable calls. During the final phase, untranslated RNAs of ribosomal and tRNA genes are annotated, and genes are classified into functional roles and color-coded for display on the now-familiar circular genome maps.
Recognizing the benefit of genome sequences to a research community that works with bacteria that are difficult to grow and manipulate, the National Institute of Dental and Craniofacial Research has funded almost all the genome projects for oral bacteria. At this writing, the genome sequences for approximately 11 oral bacteria are publicly available, and of these, four are complete (Table 1). Of the seven unfinished genomes, there is enough coverage to permit assemblies ranging from 1000 down to 100 contigs that can be downloaded from the sequencing centers for further analysis. The contigs have also been deposited at the National Center for Biotechnology Information (NCBI), where they are available for BLAST searches. Academic courtesy precludes a thorough analysis of individual genome sequences prior to publication by the sequencing team; therefore, the goal of this review is to present interesting facets of several of the publicly available sequences, and to emphasize some general concepts in comparative genomics, illustrated with relevant examples.
Porphyromonas gingivalis
Periodontal diseases have long been recognized as bacterial infections, and the presence of P. gingivalis is strongly associated with disease activity in adults. This Gram-negative anaerobe interacts with the epithelium lining the gingival sulcus, and potentially the underlying soft and hard tissues. Based on cluster and community coordination analyses, P. gingivalis was assigned to the ’red complex’ of periodontal bacteria because of associations with Treponema denticola and Bacteroides forsythus, and the positive correlation of this group with disease development (Socransky et al., 1998). Porphyromonas gingivalis is the most-studied oral bacterium, because several of its virulence factors are assayed readily, there are gene transfer systems providing the means to make mutant strains, and there are animal models for infection, as described in the comprehensive review by Holt et al. (1999).
The genome sequence of P. gingivalis strain W83 confirmed the complement of known virulence factors, e.g., gingipains and other proteinases, and genome features such as the number of ribosomal RNA operons, as well as broad aspects of P. gingivalis metabolism, notably amino acid utilization pathways (Takahashi et al., 2000), although few genes encoding peptide and amino acid transport functions have been identified thus far. From DNA-based strain-typing studies, it had long been known that, as a species, P. gingivalis appeared panmictic to the extent that only very closely related strains were groupable (Loos et al., 1993; Menard and Mouton, 1995). A possible explanation for this became apparent during sequencing and, with practical consequences, during the genome assembly phase, when a large number of potentially mobile insertion sequences (IS) had to be correctly joined to produce one contig. In strain W83, there are at least 96 complete or partial copies of IS elements that fall into 12 families. ISPg1 (formerly known as IS1126; Maley and Roberts, 1994) is the most numerous, with 36 copies, and appears to be the most active, since there was evidence of ISPg1-mediated recombination and recent transposition within W83 and other strains (Dong et al., 1999; Simpson et al., 1999). A small comparative study of the genomes of the type strain, ATCC 33277, and W83 indicated that, while gene order in the two strains was similar in at least some regions, the distribution of ISPg1 was different, strengthening the hypothesis that the species heterogeneity observed in DNA-based tests might be due to the disposition of this and other IS elements (Dong et al., 1999). Multiple copies of specific IS elements provide homology for intrachromosomal recombinations, and the resulting deletions and inversions would also generate a non-clonal population structure. These speculations were reinforced by the conclusion that ’recombination dominates over mutations in P. gingivalis’, which was derived from a study of population structures based on phylogenetic trees of housekeeping gene sequences, and the distribution of a virulence-associated gene and an IS element in a large number of clinical and laboratory strains.
Porphyromonas gingivalis was the first organism from the Cytophaga/Flavobacter/Bacteroides (CFB) phylum to be sequenced, and, not surprisingly, its sequence revealed many genes encoding orthologs also found in B. fragilis and other Bacteroides species. These include several immunoreactive surface proteins, the TonB-related protein RagA (Hanley et al., 1999), and proteins encoded by the bat operon of genes that appear to be necessary for aerotolerance, although the nature of their activities is unknown (Tang et al., 1999). In addition, there are footprints of genetic exchange with Bacteroides spp. in the form of transposon Tn5520 (Vedantam et al., 1999) and mobilization and transfer functions from the conjugative transposon CTnERL (Whittle et al., 2001; see below).
Treponema denticola
This anaerobic oral spirochete is also a member of the so-called ’red complex’ of periodontal-disease-related bacteria. Treponema denticola can be cultivated in the laboratory and is studied not only in its own right as a periodontal pathogen, but also as a relative of the syphilis spirochete, T. pallidum, that can be cultivated only in animals. Several surface proteins of T. denticola have been identified as putative virulence factors, including the major surface protein, Msp (Mathers et al., 1996; Fenno et al., 1997), and a chymotrypsin-like protease complex (Fenno et al., 1998). The potential detrimental effects of these activities on host tissues have been demonstrated with several in vitro model systems (Uitto et al., 1995; Ding et al., 1996; Wang et al., 2001). Like P. gingivalis, T. denticola uses amino acids and peptides as sources of carbon and nitrogen, and a gene cluster was identified that encoded proteins with homology to a conserved solute-binding and oligopeptide transport system, and was postulated to play a role in peptide uptake (Fenno et al., 2000). An exciting facet of T. denticola is its potential surrogacy for the culturally, hence genetically, intractable T. pallidum. Shuttle vectors were developed and introduced into T. denticola by electroporation (Li and Kuramitsu, 1996), and allelic substitution of mutant genes after electroporation of linear DNA fragments has also been demonstrated (Fenno et al., 2000). The utility of these genetic systems was demonstrated with the transfer and expression of the T. pallidum flagella gene, flaA, in T. denticola (Chi et al., 1999).
The unfinished T. denticola genome sequence is available from The Unfinished Microbial Genomes database at TIGR (www.tigr.org), and the most recent contigs were searched for open reading frames equal to or greater than 100 amino acids by means of the getORF program from the European Molecular Biology Open Software Suite (EMBOSS; Rice et al., 2000), followed by BLASTP automated searches (Altschul et al., 1997). Annotations were also compared with those of T. denticola and T. pallidum, available at the Protein Extraction, Description, and Analysis Tool (PEDANT) Web site for genome and protein analysis (http://pedant.gsf.de). These analyses of the T. denticola genome revealed that, of the closest protein homologs (n = 280, E values of 0.0 to e−116), 14% were known T. denticola genes, and, as anticipated, 64% were homologs of genes from T. pallidum Nichols, the sequenced strain. Interestingly, this homology is mostly at the protein level, with DNA sequence appearing to have diverged to greater extents than third codon wobble, reflecting their distant relationship within the Treponema genus (Paster et al., 2001). Until assembly of the T. denticola genome is complete, we will be unable to determine the extent to which gene order is conserved between T. denticola and T. pallidum; however, visual inspection of ORFs from contig assemblies shows at least four regions of significant synteny. One region of at least 16 genes is comprised of flagellar-associated functions as well as conserved hypothetical, and species-specific hypothetical, proteins. A second region of approximately 20 genes also contains hypotheticals as well as DNA primase (dnaG) and RNA polymerase sigma-70 factor adjacent to a cluster of cell-shape-determining genes (mreB, -C, and -D). A third region of 26 genes includes those encoding conserved hypothetical ORFs, cell division proteins, topoisomerase subunits, and flagellar assembly functions. Finally, a cluster of 23 genes includes genes for large and small ribosomal subunit proteins. It is not surprising that order is conserved for genes that encode essential cell functions, such as cell division and ribosome structure; however, the inclusion of ORFs for conserved and species-specific hypothetical proteins within these syntenous clusters suggests that they may also be necessary for vital cell processes, a hypothesis that is testable by mutant analysis.
In contrast to other oral bacteria, T. denticola has only four identifiable insertion sequences, three with homology to the ISL7 transposase from Lactobacillus delbrueckii (Lapierre et al., 2002), and one similar to a putative IS-like transposase from Haemophilus paragallinarum (GenBank Accession AF007429). Further, the close relative T. pallidum does not contain any insertion sequences (Fraser et al., 1998). It will be interesting to determine the extent of diversity within strains of these organisms, given that such elements have been shown to play an important role in genome plasticity and divergence in other genera (Itoh et al., 1999; Schneider et al., 2002). In this context, a small survey showed that three type strains and four clinical isolates of T. denticola were identical 16S rRNA PCR amplitypes, and a larger survey was suggested to confirm these findings (Sato and Kuramitsu, 1999).
Fusobacterium nucleatum
The first genome sequence of an oral bacterium, that of F. nucleatum strain ATCC 25586, was recently published (Kapatral et al., 2002). The strain contains a circular chromosome of 2.17 Mb with a GC content of 27%, surprisingly lower than that of most oral bacterial genomes. This feature, together with cluster analysis of orthologous genes and 16S rRNA sequence data, led to the suggestion that F. nucleatum might be grouped with the low GC Gram-positive bacteria (Kapatral et al., 2002). However, the 16S rRNA sequence assigns the organism to its own phylum within the spectrum of low GC Gram-negative bacteria (Paster et al., 2001), a placement validated by the presence of characteristic Gram-negative inner and outer cell membranes (Bolstad et al., 1996), although the latter alone does not necessarily allow for an accurate phylogenetic delineation. The higher number of orthologous clusters shared with Clostridium spp. and Bacillus subtilis, rather than with Escherichia coli, may reflect similar metabolic capacities, an issue that will be clarified by further comparisons with genomes of additional oral and non-oral anaerobes. Although ATCC 25586 does not contain plasmids, three have been identified in ATCC 10953, a second strain of F. nucleatum that is being sequenced at The Human Genome Sequencing Center, Baylor College of Medicine. These indigenous plasmids are candidate vectors for a gene transfer system (Haake et al., 2000).
A thorough analysis of the genome sequence of ATCC 25586 for metabolic pathways confirmed several earlier observations regarding the ability of F. nucleatum to use amino acids for growth (Loesche and Gibbons, 1968; Dzink and Socransky, 1990). Butyrate is a product of glutamate utilization, as with P. gingivalis (Takahashi et al., 2000), and pathways for D- and L-lysine utilization are also present in both organisms that are similar to those found in Clostridium (Chang and Frey, 2000; Ruzicka et al., 2000). Porphyromonas gingivalis and F. nucleatum can use peptides as sources of amino acids (Shah et al., 1993; Takahashi and Sato, 2002); however, in both genomes there are few candidate ORFs for peptide transporter functions. Protein localization analysis of putative peptidases from F. nucleatum ATCC 25586 predicted that the majority were located in the cytoplasm, suggesting that peptides are transported into the cell for degradation (Kapatral et al., 2002). Unlike P. gingivalis, F. nucleatum is known to take up fructose, glucose, and galactose, and phosphotransferase system transporters were found for fructose, and possibly for N-acetylglucosamine and mannose, and a glucose/galactose ABC transporter was identified similar to that found in E. coli (Kapatral et al., 2002).
Defining a role for F. nucleatum in periodontitis is confounded by the fact that the organism is a key structural component of normal and disease-associated dental plaque (Kolenbrander and Andersen, 1986; Kolenbrander, 1988; Kolenbrander and London, 1993; Suchett-Kaye et al., 1999). Fusobacteria are notable for their specific interactions with several species of oral bacteria, as illustrated by the association with Gram-positive cocci to generate characteristic ’corn-cob’ formations (Lancy et al., 1983; DiRienzo et al., 1985). Because of these properties, Fusobacteria are identified as ’co-aggregation bridge organisms’ (Kolenbrander, 2000), and attention has focused on surface proteins that might be involved in adherence and co-aggregation with other bacteria (reviewed by Bolstad et al., 1996). In the analysis of the ATCC 25586 genome sequence, outer membrane proteins received special attention as possible aggregation proteins, virulence factors, and vaccine candidates. One class of large proteins (from 188 to 362 kDa in size) was shown, on further analysis, to contain C-terminal homology to autotransporter β-domains that insert into the outer membrane, forming pores through which the associated N-terminal passenger domains, often proteases, are secreted (Duncan, unpublished; Loveless and Saier, 1997; Henderson et al., 2000). Several surface proteins of Gram-negative bacteria show autotransporter characteristics and are putative virulence factors (Pohlner et al., 1987; St. Geme and Cutter, 2000; Henderson and Nataro, 2001; Yen et al., 2002).
Prevotella intermedia
Also a member of the CFB phylum, P. intermedia forms an association complex with P. nigrescens, Fusobacterium nucleatum, and Peptostreptococcus micros (Socransky et al., 1998). Although less well-studied than P. gingivalis, several P. intermedia virulence factors have been identified, including hemagglutinating, hemolytic, and hemoglobin-binding activities (Beem et al., 1998, 1999; Leung et al., 1998; Okamoto et al., 1999). Interestingly, clinical isolates of P. intermedia from oral and extra-oral infections show resistance to antibiotics (Matto et al., 1999), and there is evidence suggesting that it is one of the more drug-resistant periodontal pathogens (Bernal et al., 1998; Fosse et al., 1999). In turn, this implies that the organism may be a reservoir for antibiotic resistance genes and a potential source of their spread among other oral species.
The unfinished P. intermedia genome sequence was obtained from The Unfinished Microbial Genomes database at TIGR, and the most recent contigs (304) were searched for open reading frames and homologies by use of the programs described above. The relationship between P. intermedia and P. gingivalis and other Bacteroides spp. in the CFB phylum was evident, since many of the predicted ORFs had homologs in these species, e.g., the bat operon of genes required for aerotolerance, and P. intermedia also contains homologs of P. gingivalis immunoreactive surface proteins and proteases. An ortholog of the P. gingivalis insertion sequence ISPg3 (formerly known as IS195; Lewis and Macrina, 1998) was found with homology at the protein and nucleotide levels, indicative of relatively recent genetic exchange between the species.
The genomes of P. intermedia and P. gingivalis have similar regions of approximately 20 kb that contain genes encoding mobilization and transfer functions of a conjugative transposon from Bacteroides; the genes and associated homologies are listed in Table 2. Analysis of the data suggests that P. intermedia and P. gingivalis versions of the transposons are related to CTnERL, since they do not contain the ermF region of CTnDOT (Whittle et al., 2001), and there is low-to-insignificant homology between their TraA-D ORFs and those encoded in CTnDOT (Bonheyo et al., 2001). It is also possible that homologies are low because parts of the region or individual genes are deleted. When the nucleotide sequences and predicted ORFs of the traE-Q genes from P. intermedia or P. gingivalis were blasted against the corresponding genes of B. thetaiotaomicron, ORFs from P. intermedia had high similarity scores, and their nucleotide sequences showed high identity over small regions, suggesting a closer or recent genetic relationship between the two sets of genes and organisms. Porphyromonas gingivalis ORFs showed lower similarities, and their nucleotide sequences showed no significant homologies. However, when the traE-Q genes and ORFs from P. intermedia were compared with those of P. gingivalis, there were greater homologies at the amino acid level and also, more significantly, at the nucleotide level across whole genes (Table 2), indicating a very close or more recent relationship. Such results raise many questions regarding conjugal transfer between organisms—for instance, Is the direction of transfer from oral to enteric anaerobes, vice versa, or in both directions? Conjugal transfer between B. thetaiotaomicron strains has been demonstrated (Li et al., 1995), and analysis of the homology data suggests relatively recent interaction between Bacteroides and P. intermedia, and between P. intermedia and P. gingivalis, but as yet there are no experimental data indicating that transfer can occur between these species. These preliminary findings call for investigations on the molecular mechanisms of DNA transfer between Bacteroides species, and it is hoped that the genome sequences will provide insights for the development of efficient gene transfer systems for P. intermedia and B. forsythus. One can then contemplate experimental approaches to determine the direction of gene transfer, the selection pressures that make transfer successful, and whether the conjugative transposons found in oral bacteria are still functional. The results will shed light on important problems regarding the use of antibiotics that may induce conjugal transposition among commensal bacteria, leading to the spread of their associated antibiotic resistance genes (Salyers and Shoemaker, 1996).
Actinobacillus actinomycetemcomitans (Aa)
A facultative oral anaerobe, Aa is strongly associated with localized juvenile periodontitis. Many virulence factors have been identified, including: surface proteins such as adhesins and invasins that mediate host tissue colonization; toxins and other proteins that promote evasion or destruction of host defenses; and factors that inhibit repair of host tissues (reviewed by Fives-Taylor et al., 1999). The sequenced strain, HK1651, is a clinical isolate, and to date the sequence is available in 88 contigs that can be downloaded from the University of Oklahoma’s Advanced Center for Genome Technology, and BLAST searches can also be carried out at NCBI. BLASTP searches of the contigs showed that, of predicted proteins with highest homologies to those from other organisms, approximately 43% have orthologs in Haemophilus influenzae, 41% in Pasteurella multocida, which causes a wide range of animal diseases, and 7.6% are known Aa proteins. The BLASTP results also showed high conservation of gene order between P. multocida and Aa, not only in gene clusters that may be putative operons, but also over large groups of non-related genes. Within some of these clusters were smaller groups of genes with synteny to the H. influenzae genome. Although these observations might suggest that Aa is more closely related to P. multocida than to H. influenzae, phylogenetic analysis based on 16S ribosomal RNA sequences indicate a closer relationship to H. influenzae. However, the high homologies shared between ORFs from the three organisms at the amino-acid and nucleotide levels clearly indicate their close relationship and relatively recent divergence.
Streptococcus mutans
Dental caries is a transmissible infectious disease in which mutans streptococci play the major role. Streptococcus mutans, the primary etiological agent, possesses several virulence factors that allow it to accumulate within the dental biofilm and produce and tolerate the acids that cause caries lesions. At this writing, the genome sequence of Streptococcus mutans is complete and will be published shortly. Of particular interest is the high degree of similarity between the genomes of S. mutans and those of S. pyogenes. Nearly half the S. mutans ORFs show highest homology to functionally similar proteins in S. pyogenes, and gene order is conserved within large regions of the genome. Although S. mutans does not contain the four complete or partial bacteriophage genome sequences that are present in S. pyogenes (Ferretti et al., 2001), it does contain numerous transposons and insertion sequences.
The S. mutans project provides the best examples of how a genome sequence can benefit the constituent basic research community. A considerable body of research is devoted to the understanding of virulence-associated acid production and acid tolerance (Quivey et al., 2001). In addition, these virulence functions are expressed in the dental plaque formed by mutans streptococci, one of the earliest-recognized microbial biofilms. Thus, a large effort is directed to an understanding of the bacterial community coordination, physiology, and life cycle inherent in biofilm formation and maintenance. A recent study took a proteomic approach to investigating the S. mutans proteins responsible for acid tolerance (Wilkins et al., 2002). Proteins in extracts of bacteria grown at pH 5.2 were compared by two-dimensional gel electrophoresis with those in extracts from bacteria grown at pH 7.0. Thirty proteins with altered expression were identified and excised, and their tryptic peptides were analyzed by matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry. The investigators used peptide mass fingerprint data to search the S. mutans database, from which 26 proteins were identified. The slow growth rate at the lower pH was reflected in the down-regulation of proteins involved in protein biosynthesis, and the up-regulation of known stress-induced proteins such as the 60-kDa chaperonin, validating the practicability of the experimental approach. Interestingly, enzymes of the glycolytic pathway were also up-regulated, and it was suggested that the accompanying increases in ATP production may result in proton efflux via H+-ATPase that contributes to aciduricity.
Functional genomic approaches were used for the identification of S. mutans genes involved in biofilm formation. Two-component signal transduction systems play an important role in bacterial sensing of, and response to, environmental changes, and homology searches detected six candidate sensor kinase-response regulator pairs in the S. mutans genome (Bhagwat et al., 2001). The response regulators were systematically mutated to assess effects on biofilm formation, and only one mutation resulted in a 10-fold reduction in biofilm growth. Ongoing studies are directed to an understanding of the environmental signals this system recognizes and the genes it regulates. Using a similar genomics-based strategy, investigators have identified an S. mutans ORF with strong homology to transcriptional regulators in other Gram-positive bacteria (Wen and Burne, 2002). A mutant defective in this gene autoaggregated in liquid cultures, formed long chains of cells, was sensitive to autolysis, and formed fragile biofilms. The pleiotrophic phenotype of the mutant suggested that the wild-type gene regulated the expression of proteins required for cell separation or autolysis that ultimately influenced biofilm formation.
Last, the comparison between S. mutans genome sequences and those of other Gram-positives has predicted models for the development of natural competence and transformation. For example, the comCDE locus, which includes the gene for the competence-inducing peptide, was identified after a search of the S. mutans genome database with the same locus from S. pneumoniae (Li et al., 2001). A further 20 proteins known to be associated with competence functions in other Gram-positives also have homologs in S. mutans, and it is anticipated that many of these will have comparable roles in transformation (discussed in the comprehensive review by Cvitkovitch, 2001).
Multiple Genome Sequences
Investigators have sequenced the genomes of different strains of several important bacterial pathogens to determine associations between different virulence phenotypes or tissue tropisms and specific sets of genes. Group A Streptococcus (GAS) shows a considerable diversity of clinical phenotypes ranging from pharyngitis to necrotizing fasciitis. GAS are classified according to the serotype of the M protein, an anti-phagocytic cell-surface protein; however, there is no association between disease phenotype and M serotype. The genome sequence of an aggressively invasive serotype M3 strain was compared with that of a serotype M1 strain that causes pharyngitis and invasive infections, and a serotype M18 strain associated with acute rheumatic fever, with the goal of identifying genes associated with a highly virulent phenotype (Beres et al., 2002). Approximately 90% of the genome was shared between the strains, and phage-related sequences accounted for the remaining 10% of diversity. The invasive M3 strain contained unique phage-encoded genes for pyrogenic toxins, a superantigen, and phospholipase activity. Similarly, as evidenced by the comparison of E. coli K12, a laboratory strain, and the virulent strain 0157:H7, lateral gene transfer, again potentially mediated by prophages, has played an important role in the acquisition of at least 130 new virulence-associated genes (Hayashi et al., 2001;Perna et al., 2001). Whole-genomic comparison of Mycobacterium tuberculosis strains H37Rv, a laboratory strain, and CDC1551, a recent clinical isolate, revealed approximately 80 large-sequence (greater than 10 bp) and over 1000 single-nucleotide polymorphisms in numerous genes (Fleischmann et al., 2002). Although previously there was thought to be little variation between the strains, the power of this high-resolution analysis demonstrated subtle differences that may have an impact on the host-pathogen interaction, since some polymorphisms were detected in genes encoding potential virulence factors and vaccine candidates.
Of the oral bacteria, F. nucleatum is the only one to date that will have complete multiple genome sequences. As stated above, the already-sequenced strain ATCC 25586 does not contain plasmids, while the second strain, ATCC 10953, contains three. Upon completion of the second sequence, it will be interesting to see the extent of strain diversity within this genus. Lack of multiple sequences has not prevented comparisons among P. gingivalis strains. Subtractive hybridization was used to identify genes that were present in the virulent sequenced strain, W83, and absent in ATCC 33277, a laboratory strain (Sawada et al., 1999). Two of the three different clones contained an insertion sequence designated IS5377 (re-named ISPg4 in the current annotation); such elements are also associated with lateral gene transfer. In lieu of sequencing multiple strains, future comparative studies can also be carried out with P. gingivalis gene microarrays (see below); however, this luxury will not be available for most oral bacteria. Should multiple strains of key organisms be sequenced? A case could be made for draft sequences, i.e., three- to six-fold coverage, which may be sufficient to identify regions of difference between virulent and avirulent strains or recent clinical isolates and laboratory-passaged strains, and may not be too costly to support.
Comparative Genomics, Lateral Gene Transfer, and Pathogenicity Islands
As stated earlier, from the comparison of multiple bacterial genome sequences, it is clear that the transfer of genes between species, i.e., lateral gene transfer, has played a major role in bacterial evolution (Doolittle, 1999). The spread of antibiotic-resistant genes between bacterial species is the most well-known manifestation of lateral gene transfer, but clusters of genes, such as those encoding a metabolic pathway, may also be transferred as a functional unit to a new host if they confer an ecological advantage, as proposed in the selfish operon model (Lawrence and Roth, 1996). Whether a gene has the potential for successful transfer appears to depend on its function. Scatterplots based on sequence-similarity scores of orthologous gene sets from several prokaryote genomes indicated that genes fell into two groups: informational genes that function in translation, transcription, and replication; and operational genes that encode enzymes in metabolic pathways, and regulatory functions (Rivera et al., 1998). Further phylogenetic analyses of over 300 orthologous genes from six bacterial genomes established that operational genes had been, and still were, extensively transferred, while informational genes were transferred rarely (Jain et al., 1999). According to the complexity hypothesis proposed to account for these findings, the product of an informational gene, such as an enzyme, has a small number of interactions, e.g., with its substrate and perhaps a co-factor; therefore, transfer of the coding gene to a new host will result in relatively few compensating adjustments to ensure working efficiency and host survival. In contrast, a ribosomal protein may interact with four or five other ribosomal components; thus, transfer of its coding gene to a new host may involve extensive accommodations that require compensating mutations in genes for other ribosomal products, making the chances of successful transfer, as manifested by the viability of the new host, relatively small.
Clues suggesting that a gene may have been acquired by lateral gene transfer include a GC content and/or codon usage different from those of the other host genes, antibiotic-resistant functions, activities associated with virulence, and genetic linkage with known moveable DNA elements. Many of these criteria are fulfilled by pathogenicity islands, so-called because they contain genes for virulence factors in micro-organisms that cause disease (reviewed by Hacker and Kaper, 2000). Ranging in size from 10 to 200 kb, pathogenicity islands often carry genes encoding integrases and transposases that are involved in DNA mobility, and they may be associated with transfer RNA genes, favored sites for the integration of foreign DNA. Are there examples of pathogenicity islands in the genomes of oral bacteria? A 24-kb region of the Aa genome was the first pathogenicity island to be characterized in an oral pathogen (Mayer et al., 1999). It carries a characterized virulence gene locus, that of the cytolethal distending toxin, a bacteriophage attachment site, and an integrase/resolvase. More recent results indicate that the region is flanked by 160-bp repeats and contains a total of 19 ORFs (Doungudomdacha and DiRienzo, manuscript in preparation). In the P. gingivalis genome, there are two paralogous regions that have all the hallmarks of pathogenicity islands, one of approximately 28 kb, and a deleted version of approximately 18 kb (Fig. 2). It is probable that, after the initial putative transfer, the paralogs were generated by duplication and intrachromosomal recombination. With an average G+C composition of 41%, compared with a 48% average for the whole genome, the regions are bound on one side by homologs of Bacteroides transposon Tn5520, and on the other by either a serine or aspartate tRNA. Half the ORFs encode homologs of transcription regulators, mobilization and transfer functions of Bacteroides conjugative transposons, excisases and integrases, ISPg1, and an efflux pump family protein. The other ORFs encode either conserved hypothetical or species-specific hypothetical ORFs, and the challenge of identifiying the functions of unknown ORFs within these regions will determine whether they are true pathogenicity islands. The existence of these atypical islands again prompts the question of how they got there. Clearly, the close and constant bacterial associations in dental plaque present favorable conditions for the transfer of conjugative transposons by cell-to-cell contact (Roberts et al., 1999; Waters, 1999), and recently it has been shown that natural competence for DNA uptake increases when bacteria are grown in plaque-like biofilms (Li et al., 2001, 2002; Wang et al., 2002).
Convergent Evolution?
In several of the Gram-negative oral anaerobes, there is a class of proteins that contains leucine-rich repeats (LRR). The first such sequence to be published was of a B. forsythus surface component, BspA (Fig. 3A), a 98-kDa protein that contains 14 tandem arrays of 23 amino acids (Sharma et al., 1998). Subsequently, a P. gingivalis protein of 145 kDa was identified with at least 16 tandem repeats of 22 amino acids (Fig. 3A; Yong et al., 2000) that shared homology with internalin A from Listeria monocytogenes (Gaillard et al., 1991). LRRs are present in many proteins of both eukaryotes and prokaryotes, and are associated with diverse functions including transmembrane receptors, cell adhesion, and signal transduction, where the LRR protein recognition motif provides a framework for protein-protein interactions (Buchanan and Gay, 1996). Genes for BspA-like proteins were also found in the B. fragilis, T. denticola, and P. intermedia genomes, and internalin-like variants were found in P. gingivalis and P. intermedia. Antibody staining of BspA in B. forsythus (Sharma et al., 1998) showed the location of the protein to be extracellular. LRRs from other organisms contained either predicted signal peptides and/or bacterial lipopolysaccharide attachment motifs, also indicative of cell-surface proteins. The presence of the cysteine residues at the C-terminus of each repeat, exemplified with BspA in Fig. 3A, also denotes an extracellular location (Buchanan and Gay, 1996). The larger internalin-like protein of P. gingivalis shows another structural variation in that cysteine residues flank the LRR domain instead of being part of the sequence. The cysteine residues are thought to play a structural role by forming disulphide bonds within the predicted extracellular regions. In many LRR proteins, the repeat regions form β-sheet structures, with the functional residues facing outward for protein-protein interactions, while the specificity of these interactions may reside in the unique N- and C-termini of the proteins. As yet, only the protein from B. forsythus has been assigned a function in adherence to extracellular matrix proteins (Sharma et al., 1998), and similar functions may also be predicted for the P. gingivalis, P. intermedia, and T. denticola proteins.
There is no evidence of the spread of LRRs within prokaryotes by lateral gene transfer, and it is not known whether they share a common ancestor. The LRR motif and subsequent duplications may have occurred independently many times, and perhaps represents a case of convergent evolution whereby increased protein-binding capacity due to multiple tandem repeats is the most advantageous structure. It has also been suggested that the binding surface of prokaryotic LRRs mimics that of eukaryotic receptors, and was cited as an example of molecular mimicry in which a cellular-receptor binding module is subverted by micro-organisms for pathogenic purposes (Stebbins and Galan, 2001).
Influence of Bacterial Genomics on Basic and Clinical Research
The availability of genome sequences has changed the way laboratories, large and small, carry out research in bacterial genetics. Freed from the obligation and tedium of sequencing, investigators can move on to the more interesting topics of gene function and regulation. And there is plenty of work ahead, since, even in well-studied bacteria, approximately 25% of genes code for species-specific proteins of unknown function, illustrating the extraordinary amount of biological diversity that previously we did not know existed. Speculations as to the roles of these newly discovered genes range from regulators to activities required for survival in animal hosts. Understanding the molecular mechanisms of bacterium-host interactions is central to research in microbial pathogenicity and infectious disease. These interactions take place in complex and dynamic in vivo environments, and to be a successful colonizer of host tissue, a bacterium must interact with other micro-organisms, compete for nutrients, and ward off the challenge of host defenses, conditions that are impossible to simulate in vitro. Several in vivo expression technologies (IVET) use animal models to identify genes that are up-regulated or expressed only in vivo (reviewed by Chiang et al., 1999). The more recently developed in vivo-induced antigen technology (IVIAT) uses patient sera rather than animals to probe for proteins, thus identifying genes that are specifically induced during human infection (Handfield et al., 2000). An IVET system was used to identify P. gingivalis genes expressed in a mouse abscess model, and of the 14 genes published, 50% were functionally unknown ORFs (Wu et al., 2002). For the comparatively less-well-studied oral pathogens, the availability of genome sequences makes these studies practicable, since often only fragments of the in vivo-induced genes are represented in the expression libraries; however, even from small amounts of sequence, complete genes can be retrieved from genome databases. In addition, when sequences from hypothetical genes are identified, the database provides some validation that they are genuine, and their genetic context can provide clues as to function. The recently discovered associations of specific bacteria with systemic diseases, e.g., Helicobacter pylori as the etiological agent of gastric ulcers, drive new research initiatives to assess the association of P. gingivalis, and other oral pathogens also detected in atherosclerotic plaques (Chiu, 1999; Shelburne et al., 2000), with cardiovascular disease (Herzberg and Weyer, 1998; Meyer and Fives-Taylor, 1998; Offenbacher et al., 1999; Scannapieco and Genco, 1999). These studies suggest that the pathogenicity of oral bacteria may extend beyond their known ecological niche to other organ systems, introducing an important and exciting new dimension to defining the molecular mechanisms of pathogenesis.
A completed genome sequence also places an organism in the new and exciting realm of functional genomics. Our collaborators at TIGR generated PCR amplicons of all P. gingivalis ORFs and printed microarrays (DNA chips) for whole-genome expression profiling. Thus, the organism is now more accessible for study, not only for its virulence potential, but also for other aspects of physiology that relate to pathogenesis, such as: the regulation of virulence factors; effects of drugs, their metabolism, and resistance mechanisms; identification of functionally unknown genes by virtue of their co-regulation; how mutations affect gene expression; and host-induced and -repressed activities. The principle behind microarray technology is DNA-DNA hybridization in a dot-blot format (Schena et al., 1995). DNA sequences from each gene are spotted robotically, in known order, onto an impermeable matrix, usually glass. Total RNA is isolated from bacteria grown under an experimental and control growth condition, e.g., with and without antibiotic. Messenger RNA is reverse-transcribed to cDNA by random hexamers, and specific fluorescent dyes are conjugated to the experimental and control cDNA libraries for differentiation. The cDNA libraries are mixed and incubated with the arrayed genes (probes), and control and experimental hybridizations are detected with scanning lasers that recognize the different fluorescent dyes. The expression value of a specific gene is calculated from the ratio of the dyes bound to its DNA probe. Because microarray analysis identifies all the genes induced by the stimulus, a comprehensive profile is obtained of the extent of the response in co-regulated pathways, and the phrase ’global analysis of gene expression’ aptly describes the results.
Genome information is being utilized to design new vaccines, and one approach, called ’reverse vaccinology’ (Rappuoli, 2001), was used to identify new vaccine targets in serogroup B Neisseria meningitidis (Pizza et al., 2000). The genome sequence was analyzed in silico for proteins with cell-surface or secreted protein signatures, such as signal sequences, transmembrane domains, homology to known surface proteins, and lipoprotein, outer membrane anchor, and RGD motifs. Nearly 600 putative proteins were identified, and more than half of these could be expressed in E. coli. The recombinant proteins were used to immunize mice, and immune sera were tested for bactericidal activity and immunoreactivity to whole bacteria. Out of the seven candidates that passed these screens, two conserved proteins are now in clinical trials as potential protective immunogens. Clearly, this strategy has several advantages over conventional approaches for identifying vaccine candidates, the most obvious being the speed of the initial identification phase. In addition, it offers the possibility of finding targets that are expressed only in vivo and, more importantly, may be applicable to non-cultivable organisms.
New Challenges: Uncultivable Bacteria and Microbial Communities
Most of the micro-organisms whose genomes have been sequenced so far can be grown in the laboratory, albeit sometimes under complex conditions, such as in epithelial cells for Chlamydia spp. However, it is estimated that greater than 99% of the micro-organisms observed in nature cannot be cultivated by standard techniques (Amann et al., 1995). This holds true for the oral flora, since, despite significant efforts, only about 50% of the approximately 500 species of bacteria present in the oral cavity are cultivable (Paster et al., 2001). With this knowledge, coupled with the fact that, in nature, most bacteria live in communities, one can see that genome sequencing is poised to undertake two new challenges: to sequence the genome of a specific uncultivable bacterium; and to sequence the ’metagenome’ of a community of cultivable and non-cultivable bacteria from a specific niche. The oral microbiota is the ideal community for meeting both challenges. Following the methods established in a pioneering study to investigate the microbial diversity or metagenome of soil (Rondon et al., 2000), health- or disease-associated dental plaque would be the source of genomic DNA for both an uncultivable organism and the microbial consortium. In both cases, the DNA must be handled carefully to preserve its integrity and maximize the chances of cloning large stretches of genetic information, and fractionation by pulsed field gel electrophoresis has been used successfully to isolate such large intact fragments. The first phase in the conventional random shotgun sequencing strategy for a single bacterial genome involved making a library of small genomic fragments of 2 kb. A metagenome may be orders of magnitude larger, requiring an extra step to clone large DNA fragments into bacterial artificial chromosome vectors (BACs) that can maintain up to 100-kb inserts. BAC inserts can then be treated like mini-genomes and mechanically sheared to generate small fragment libraries and used for end-sequencing and assembly, as for a single genome. Constituent organisms of the metagenome can be identified after amplification and cloning of 16S ribosomal RNA fragments, followed by database searching with individual sequences. The uncultivable bacterium can be identified from its 16S ribosomal RNA sequence, and the BAC insert containing this gene would be the ’seed’ fragment. The end sequences of the insert can be used to probe a BAC library for clones that contain overlapping DNA, and by this iterative process as much of the genome as possible retrieved.
Conclusions
Without doubt, genome sequencing has had broad and revitalizing impacts on all aspects of microbiology. Besides providing relief from the gene-by-gene sequencing grind, it has enhanced, and sometimes confounded, our understanding of bacterial evolution, expedited research in microbial pathogenesis, and opened new vistas in protein chemistry with the discovery of hundreds of unknown proteins and unique folding patterns. Oral microbiology has been energized by, and will continue to benefit from, the genome projects of its constituent organisms, and those of close relatives among the non-oral pathogens. Working with oral bacteria that grow slowly, have complex growth requirements, and lack gene transfer systems is both challenging and frustrating. The genome sequences of refractory oral organisms will provide insights into their metabolism that will lead to the development of better growth conditions, and evidence of past genetic exchanges will stimulate new gene transfer strategies. New projects to sequence microbial consortia or uncultivable organisms will also benefit and widen the horizons of our research community.
While this paper was under review, the genome sequence of S. mutans UA 159 was published by Ajdic et al.(2002). The genome of Bacteroides thetaiotaomicron, recently published by Xu et al.(2003), is more useful for comparative analyses of P. gingivalis genes.
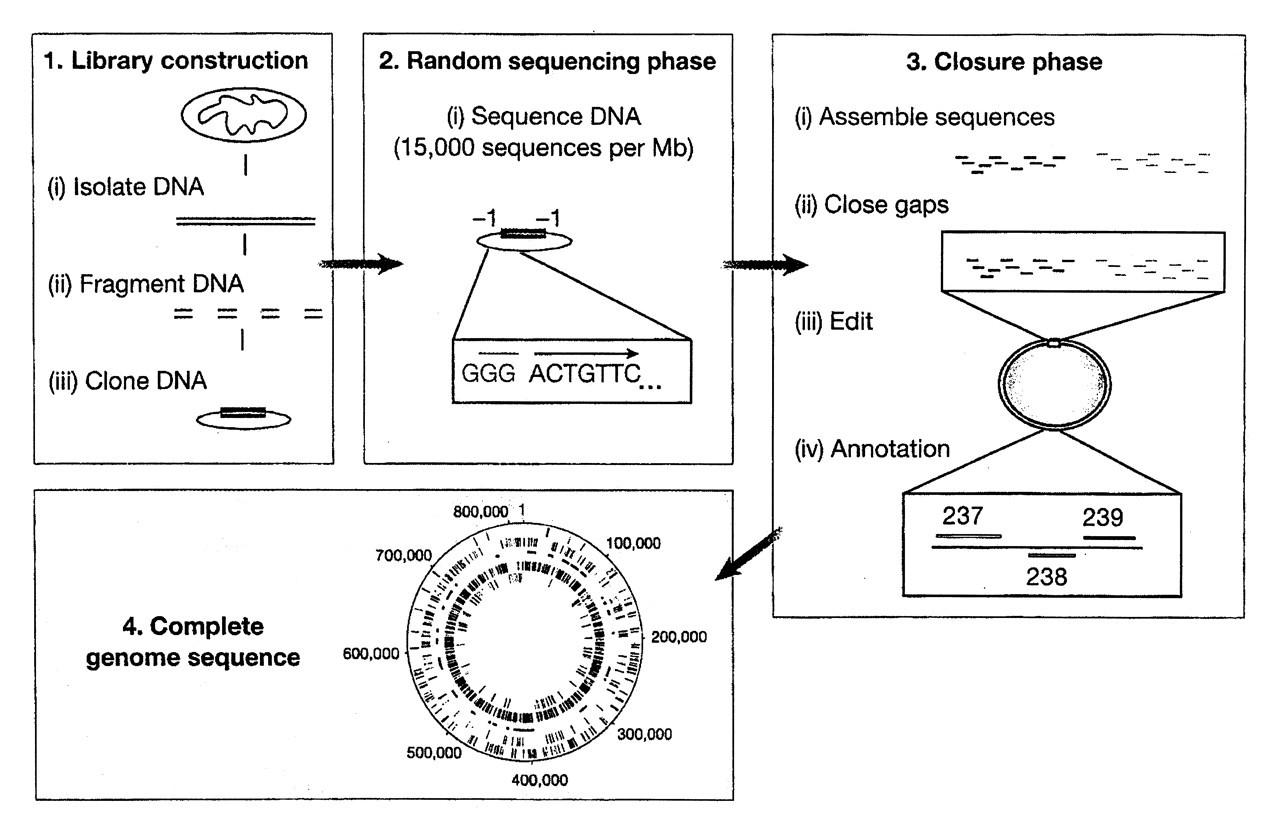
The four phases of a whole-genome sequencing project. From Fraser et al. (2000); reprinted with permission.
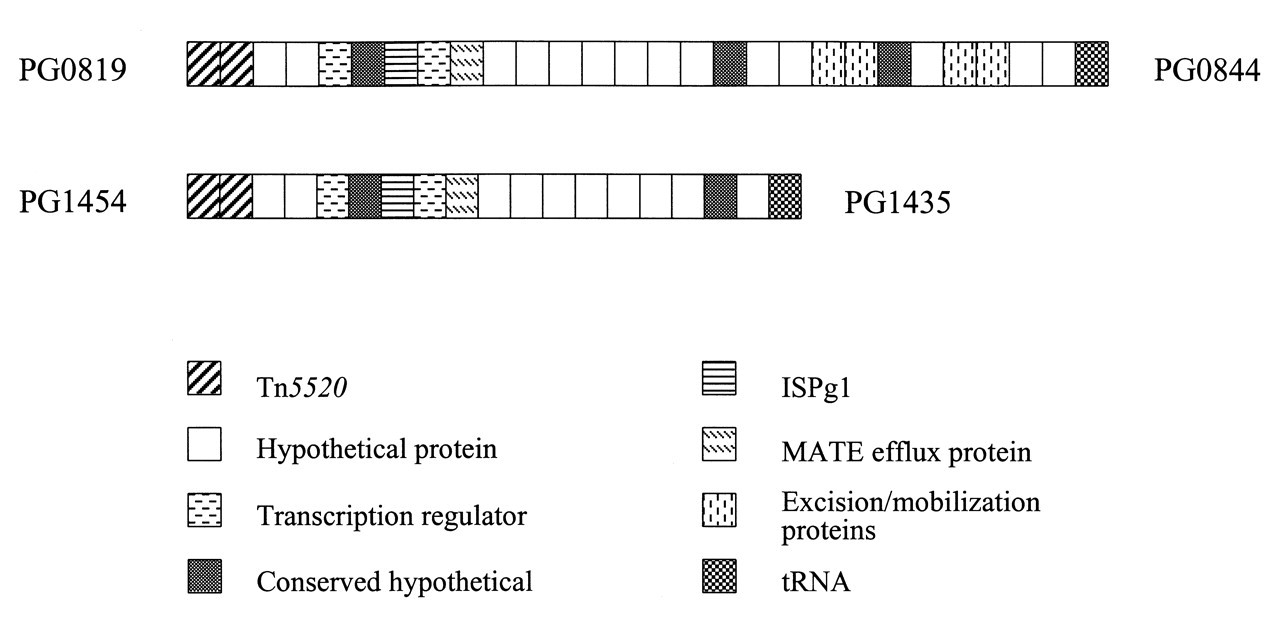
Putative pathogenicity islands in the P. gingivalis genome. PG numbers are given for the first and last genes in each region. The paralogous regions are approximately 28 and 18 kb. Functional assignments are conditional and were obtained from the TIGR and LANL annotations.

Leucine-rich repeats in putative surface proteins of oral bacteria.
Footnotes
Acknowledgements
Thanks are due to the NIDCR for funding the genome projects for oral bacteria, and especially to Dr. Dennis Mangan, the champion of the cause. Work on the P. gingivalis genome was supported by NIH grants DE 12082 (R.D. Fleischmann) and DE 10510 (M.J.D.). I thank Dr. T. Chen for setting up the automated annotation programs, and Drs. B. Paster and D. Fraenkel for helpful discussions.