
Abstract
Both antigen-specific and non-specific mechanisms may be involved in the pathogenesis of oral lichen planus (OLP). Antigen-specific mechanisms in OLP include antigen presentation by basal keratinocytes and antigen-specific keratinocyte killing by CD8+ cytotoxic T-cells. Non-specific mechanisms include mast cell degranulation and matrix metalloproteinase (MMP) activation in OLP lesions. These mechanisms may combine to cause T-cell accumulation in the superficial lamina propria, basement membrane disruption, intra-epithelial T-cell migration, and keratinocyte apoptosis in OLP. OLP chronicity may be due, in part, to deficient antigen-specific TGF-β1-mediated immunosuppression. The normal oral mucosa may be an immune privileged site (similar to the eye, testis, and placenta), and breakdown of immune privilege could result in OLP and possibly other autoimmune oral mucosal diseases. Recent findings in mucocutaneous graft-versus-host disease, a clinical and histological correlate of lichen planus, suggest the involvement of TNF-α, CD40, Fas, MMPs, and mast cell degranulation in disease pathogenesis. Potential roles for oral Langerhans cells and the regional lymphatics in OLP lesion formation and chronicity are discussed. Carcinogenesis in OLP may be regulated by the integrated signal from various tumor inhibitors (TGF-β1, TNF-α, IFN-γ, IL-12) and promoters (MIF, MMP-9). We present our recent data implicating antigen-specific and non-specific mechanisms in the pathogenesis of OLP and propose a unifying hypothesis suggesting that both may be involved in lesion development. The initial event in OLP lesion formation and the factors that determine OLP susceptibility are unknown.
Introduction
Oral lichen planus (OLP) is a T-cell-mediated chronic inflammatory oral mucosal disease of unknown etiology. OLP lesions contain few B-cells or plasma cells and minimal deposits of immunoglobulin or complement. There are no consistent serological changes associated with OLP. The oral mucosa in OLP is highly accessible for detailed investigation. Hence, OLP is ideally positioned for the study of human T-cell-mediated inflammation and autoimmunity. Our research over the past 10 years has focused on three basic questions: (i) Why and how do T-cells accumulate in the superficial lamina propria in OLP? (ii) Why and how do T-cells enter the oral epithelium in OLP? and (iii) What triggers basal keratinocyte apoptosis in OLP? In our attempts to answer these questions, it became apparent that both antigen-specific and non-specific mechanisms might be involved. We now present our data that led to this conclusion and propose a unifying hypothesis for the pathogenesis of OLP.
Clinical Features of OLP
OLP presents as white striations, white papules, white plaques, erythema, erosions, or blisters affecting predominantly the buccal mucosa, tongue, and gingivae (Vincent et al., 1990; Silverman et al., 1991). OLP affects one to two percent of the general adult population and is the most common non-infectious oral mucosal disease in patients referred to Oral Medicine and Oral Pathology clinics (Axéll and Rundqvist, 1987; Bowers et al., 2000). OLP affects women more than men at a ratio of approximately 1.4:1 (Axéll and Rundqvist, 1987). OLP occurs predominantly in adults over 40, although younger adults and children may be affected (Vincent et al., 1990; Chainani-Wu et al., 2001). Lesions are usually bilateral, and atrophic and erosive lesions are often sensitive or painful (Scully and El-Kom, 1985; Eisen, 1993). There may be co-incident skin lesions that present typically as flat-topped violaceous papules affecting the wrists, ankles, and genitalia. Nail involvement results in pitting, pterygium formation, and permanent nail loss. Scalp involvement results in scarring alopecia (Sugerman et al., 2000a). Rarely, there is laryngeal, esophageal, and conjunctival involvement (Eisen, 1999). There is ongoing concern that OLP may be pre-malignant, although the malignant transformation data are currently under review, and further prospective studies are required. In patients who do not use tobacco products, squamous cell carcinoma (SCC) may arise at the site of a pre-existing OLP lesion in less than five percent of cases, most frequently in atrophic, erosive, and plaque lesions. Hence, OLP patients are at slightly increased risk of oral cancer, although it is unlikely that OLP is inherently pre-malignant (Eisenberg, 2000; Silverman, 2000).
Oral mucosal lichenoid lesions sometimes follow (with a variable lag period) the administration of a systemic drug. These lichenoid drug reactions may be unilateral but otherwise appear as idiopathic OLP. Drugs that have been implicated in oral lichenoid drug reactions include non-steroidal anti-inflammatory drugs, angiotensin-converting enzyme inhibitors, and beta-blockers, although there are many others (Porter and Scully, 2000). Oral mucosal lichenoid lesions may follow the placement of a dental restoration or provision of a denture, again with a variable lag period. These lichenoid contact sensitivity reactions are usually in contact with (or close to) an amalgam or composite resin dental restoration or a denture component (Lind, 1988; Bolewska et al., 1990). Flavorings, especially cinnamates in toothpaste, may also trigger lichenoid contact sensitivity reactions, although there is little published evidence in support of this observation (Miller et al., 1992; Yiannias et al., 2000). Circumstantial evidence implicates various bacteria and viruses in the etiology of OLP, although any causal role remains speculative (Scully et al., 1998).
Histology of OLP
The histology of OLP is characterized by a dense subepithelial lympho-histiocytic infiltrate, increased numbers of intra-epithelial lymphocytes, and degeneration of basal keratinocytes. Degenerating basal keratinocytes form colloid (Civatte, hyaline, cytoid) bodies that appear as homogenous eosinophilic globules. The ultrastructure of colloid bodies suggests that they are apoptotic keratinocytes, and recent studies using the end-labeling method demonstrated DNA fragmentation in these cells (Hashimoto, 1976; Weedon, 1980; Dekker et al., 1997; Shimizu et al., 1997; Bloor et al., 1999; Neppelberg et al., 2001). Epithelial basement membrane changes are common in OLP and include breaks, branches, and duplications (Jungell et al., 1989a; Zhou et al., 2001). In addition, the basal keratinocyte anchoring elements (hemidesmosomes, filaments, and fibrils) are disrupted in OLP (Haapalainen et al., 1995). Degeneration of basal keratinocytes and disruption of the epithelial basement membrane and basal keratinocyte anchoring elements in OLP produce weaknesses at the epithelial-connective tissue interface which may result in histological cleft formation (Max-Joseph space) and, rarely, clinical blistering of the oral mucosa (bullous lichen planus). Parakeratosis, acanthosis, and “saw-tooth” rete peg formation may be seen. B-cells and plasma cells are infrequent in OLP, and immunoglobulin and complement deposits are not a consistent feature. Some cases show fibrinogen and fibrin deposition in a linear pattern in the basement membrane zone. Colloid bodies may be positive for fibrin, IgM, C3, C4, and keratin. Laminin and fibronectin staining may be absent in areas of heavy fibrin deposition and colloid body formation, suggesting basement membrane damage in these areas. Immunofluorescent findings in OLP are not diagnostic (Scully et al., 1998).
Antigen Specificity in OLP
CD8+ T-cells
The lymphocytic infiltrate in OLP is composed almost exclusively of T-cells, and the majority of T-cells within the epithelium and adjacent to damaged basal keratinocytes are activated CD8+ lymphocytes (Matthews et al., 1984; Kilpi, 1987, 1988; Jungell et al., 1989b). In our recent studies, the majority of subepithelial and intra-epithelial lymphocytes in OLP were CD8+ (Khan et al., 2001, submitted). We also observed CD8+ T-cells that co-localized with apoptotic keratinocytes in OLP lesions (Sugerman et al., 2000a; Khan et al., 2001, submitted). Analysis of these data suggests that CD8+ T-cells are involved in disease pathogenesis, and that activated CD8+ T-cells may trigger keratinocyte apoptosis in OLP. T-cell lines and clones isolated in vitro from lichen planus lesions were more cytotoxic against autologous lesional keratinocytes than T-cell lines and clones from the clinically normal skin of lichen planus patients (Sugerman et al., 2000b). Lesional T-cell clones were more cytotoxic against autologous lesional keratinocytes and normal skin keratinocytes than against autologous B-cell blasts. The majority of cytotoxic clones from lichen planus lesions were CD8+, and the majority of non-cytotoxic clones were CD4+. The cytotoxic activity of CD8+ lesional T-cell clones was partially blocked with anti-MHC class I monoclonal antibody (Sugerman et al., 2000b). Hence, early in OLP lesion formation, CD8+ lesional T-cells may recognize an antigen associated with MHC class I on lesional keratinocytes. Following antigen recognition and activation, CD8+ cytotoxic T-cells may trigger keratinocyte apoptosis. T-cell activation and subsequent clonal expansion may underlie restricted T-cell receptor Vβ gene expression (especially Vβ22 and Vβ23) by infiltrating T-cells in OLP (Zhou et al., 1996). Activated CD8+ T-cells (and possibly keratinocytes) may release chemokines that attract additional lymphocytes and other immune cells into the developing OLP lesion (Yamamoto et al., 1994; Yamamoto and Osaki, 1995). In cutaneous lichen planus, a dermal infiltrate of T-lymphocytes preceded histological epithelial damage. In slightly more advanced lesions, lymphocytes were seen within the lower epidermis, and at this stage, there was evidence of epithelial damage, including vacuolar alteration of basal keratinocytes and slight spongiosis in the spinous zone (Ragaz and Ackerman, 1981). These findings are consistent with the current hypothesis that intra-epithelial CD8+ T-cells trigger keratinocyte apoptosis in OLP.
Initial approximation between CD8+ T-cells and keratinocytes
Previous studies identified various biochemical changes associated with altered keratinocyte antigen expression and altered keratinocyte function early in lichen planus lesion formation (Black and Wilson-Jones, 1972; Heyden et al., 1974; Holmstrup and Dabelsteen, 1979). The authors suggested that these epithelial changes were initial events in disease pathogenesis. Following altered keratinocyte antigen expression, an antigen-specific CD8+ T-cell may be either (i) on routine surveillance in the epithelium and encounter the keratinocyte antigen by chance (“chance encounter” hypothesis) or (ii) attracted to the epithelium, along with T-cells of irrelevant specificity, by keratinocyte-derived chemokines (“directed migration” hypothesis). The “chance encounter” hypothesis is supported by findings of CD8+ T-cells in normal human epidermis (Bos et al., 1987; Spetz et al., 1996) and basal cell degeneration in the absence of a dense inflammatory infiltrate in lichen planus lesions (Sarkany and Gaylarde, 1971). The “chance encounter” hypothesis may explain the prevalence of OLP in the general population (one to two percent) and the onset of OLP in later life, i.e., it takes some time for the CD8+ T-cell to encounter its specific antigen in the oral epithelium. Conversely, the “directed migration” hypothesis is supported by findings of constitutive chemokine receptor expression on naïve T-cells (Sallusto et al., 1998) and a dermal T-cell infiltrate prior to the appearance of intra-epithelial lymphocytes and epithelial damage in lichen planus lesions (Ragaz and Ackerman, 1981). In addition, pre-activation of antigen-specific CD8+ T-cells (e.g., in a regional lymph node) may up-regulate inflammatory chemokine receptor expression and facilitate antigen-specific CD8+ T-cell migration to the future OLP lesion site (Walsh et al., 1990b). The “directed migration” hypothesis predicts that keratinocyte-derived chemokines attract T-cells of irrelevant specificity along with antigen-specific T-cells into the developing OLP lesion. In support of this hypothesis, our recent studies identified a significant proportion of non-clonal OLP lesional T-cells (Zhou et al., 1996), and not all CD8+ lesional T-cell clones were cytotoxic against autologous lesional keratinocytes in vitro (Sugerman et al., 2000b). In summary, the initial event in OLP lesion formation may be keratinocyte antigen expression in association with MHC class I at the future lesion site, with or without up-regulated keratinocyte chemokine production. Pioneer antigen-specific CD8+ cytotoxic T-cells may enter the oral epithelium on routine surveillance, or they may be attracted by keratinocyte-derived chemokines. Subsequently, antigen-specific CD8+ cytotoxic T-cells trigger apoptosis of basal keratinocytes. In this context, keratinocyte antigen expression and chemokine production are primary events in OLP lesion formation, followed by keratinocyte apoptosis triggered by antigen-specific CD8+ cytotoxic T-cells. Formation of the dense subepithelial lympho-histiocytic infiltrate and epithelial basement membrane changes in OLP may result from antigen-specific interactions between keratinocytes and T-cells, or they may be epiphenomena associated with the recruitment of T-cells with irrelevant specificity into the OLP lesion site. The roles of keratinocyte-derived chemokines and lymphocyte chemokine receptor expression in OLP lesion formation are currently under investigation.
Identity and location of the lichen planus antigen
The lichen planus antigen is unknown, although the antigen may be a self-peptide, thus defining lichen planus as a true autoimmune disease. The role of autoimmunity in disease pathogenesis is supported by many autoimmune features of OLP, including disease chronicity, adult onset, female predilection, association with other autoimmune diseases, occasional tissue-type associations, depressed immune suppressor activity in OLP patients, and the presence of auto-cytotoxic T-cell clones in lichen planus lesions (Sugerman et al., 1992, 1993, 2000b). We suggest that keratinocytes express a lichen planus antigen but only at the lesion site, i.e., the clinical distribution of lichen planus lesions is determined by the distribution of the lichen planus antigen. Hence, an early event in lichen planus lesion formation may be keratinocyte antigen expression or unmasking at the future lesion site induced by systemic drugs (lichenoid drug reaction), contact allergens in dental restorative materials or toothpastes (contact hypersensitivity reaction), mechanical trauma (Koebner phenomenon), bacterial or viral infection, or an unidentified agent. Subsequently, intra-epithelial CD8+ cytotoxic T-cells recognize the lichen planus antigen associated with MHC class I on lesional keratinocytes and trigger keratinocyte apoptosis.
Heat-shock proteins
We and others identified up-regulated heat-shock protein (HSP) expression by OLP lesional keratinocytes in situ (Bramanti et al., 1995; Sugerman et al., 1995; Chaiyarit et al., 1999). We also identified HSP-reactivity of OLP lesional T-cells in vitro (Sugerman et al., 1995). Keratinocyte HSP expression in OLP may be an epiphenomenon associated with pre-existing inflammation. Alternatively, up-regulated HSP expression by oral mucosal keratinocytes may be a common final pathway linking a variety of exogenous agents (systemic drugs, contact allergens, mechanical trauma, bacterial or viral infection) in the pathogenesis of OLP. In this context, HSP expressed by oral keratinocytes may be auto-antigenic in OLP (Sugerman et al., 1995). Susceptibility to OLP may result from dysregulated HSP gene expression by stressed oral keratinocytes or from an inability to suppress an immune response following self-HSP recognition. The latter mechanism seems likely, given our finding of depressed immunological suppressor activity in OLP patients (Sugerman et al., 1992). However, in vitro examination of the kinetics of HSP expression by heat-shocked oral mucosal keratinocytes in OLP patients may reveal an alternative or additional explanation for OLP susceptibility. Of further interest, both murine (Bensaude and Morange, 1983; Ullrich et al., 1986) and human (Fisch et al., 1990; Kaur et al., 1993) transformed cell lines and human malignant lesions (Ciocca et al., 1993; Kimura et al., 1993) express constitutively high levels of HSP. Hence, some cases of malignant transformation of OLP lesions may actually represent an initial lichenoid response to pre-existing tumor cells expressing elevated levels of HSP, followed by the development of overt clinical and histological malignancy.
Mechanisms of keratinocyte killing
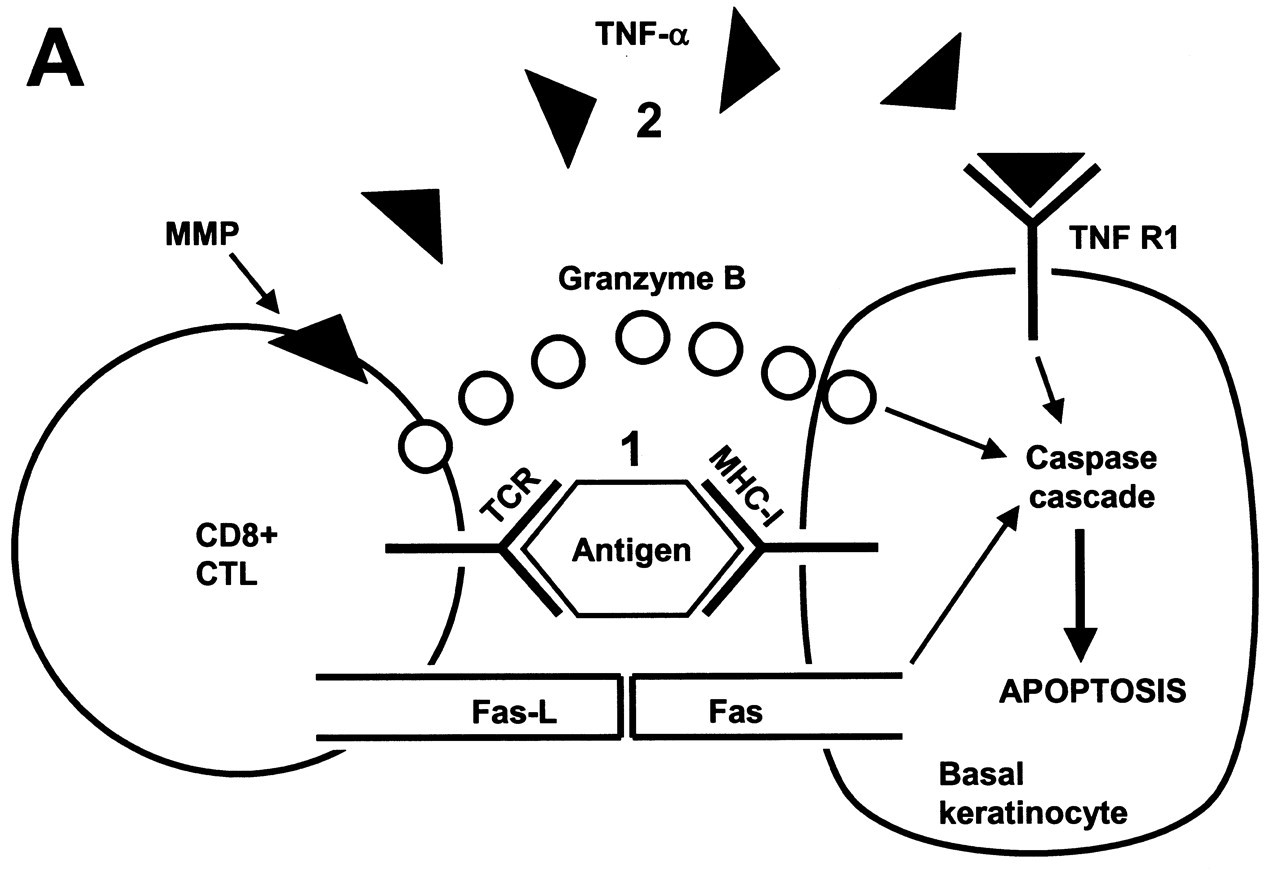
Currently, the mechanisms used by CD8+ cytotoxic T-cells to trigger keratinocyte apoptosis in OLP are unknown. Possible mechanisms include (i) T-cell-secreted TNF-α binding TNF-α receptor 1 (TNF R1) on the keratinocyte surface, (ii) T-cell surface CD95L (Fas ligand) binding CD95 (Fas) on the keratinocyte surface, or (iii) T-cell-secreted granzyme B entering the keratinocyte via perforin-induced membrane pores. All of these mechanisms may activate the keratinocyte caspase cascade, resulting in keratinocyte apoptosis (Sugerman et al., 2000a). We identified elevated levels of TNF-α in the serum of OLP patients and showed that OLP lesional T-cells contained mRNA for TNF-α and secreted TNF-α in vitro (Sugerman et al., 1996; Simark-Mattsson et al., 1999; Khan et al., 2001, submitted). TNF-α was expressed by T-cells throughout the subepithelial infiltrate and by T-cells adjacent to basal keratinocytes in OLP lesions. The TNF-α receptor TNF R1 was expressed by basal and suprabasal epithelial cells in OLP lesions (Khan et al., 2001, submitted). Hence, T-cell-secreted TNF-α may be involved in the pathogenesis of OLP. CD8+ cytotoxic T-cells in OLP may secrete TNF-α that triggers keratinocyte apoptosis via TNF R1.
CD4+ T-cells
While the majority of intra-epithelial lymphocytes in OLP are CD8+ cytotoxic T-cells, most lymphocytes in the lamina propria are CD4+ helper T-cells (Matthews et al., 1984; Ishii, 1987; Kilpi, 1988). Our previous studies identified mixed helper and suppressor activity among OLP lesional T-cell clones in vitro, suggesting that the balance between immunological help and suppression may determine the clinical behavior of the disease (Sugerman et al., 1994). We also isolated non-cytotoxic CD4+ T-cell clones from cutaneous lichen planus lesions in vitro (Sugerman et al., 2000b). Hence, an early event in OLP lesion formation may be MHC class II antigen presentation to CD4+ helper T-cells, followed by keratinocyte apoptosis triggered by CD8+ cytotoxic T-cells. MHC class II antigen presentation in OLP may be mediated by Langerhans cells (LCs) or keratinocytes. There are increased numbers of LCs in OLP lesions with up-regulated MHC class II expression (Rich and Reade, 1989; Farthing et al., 1990). Keratinocytes in OLP also express MHC class II (Farthing and Cruchley, 1989; Walsh et al., 1990a). High levels of antigen expression, CD40 and CD80 expression, and IL-12 secretion by MHC class II+ antigen-presenting cells (APC) in OLP may promote a T-helper-1 (Th1) CD4+ T-cell response with IL-2 and IFN-γ secretion (Constant and Bottomly, 1997). In support of this hypothesis, human epidermal LCs and keratinocytes are capable of producing IL-12 (Muller et al., 1994; Kang et al., 1996). In addition, we identified IFN-γ expression by T-cells adjacent to basal keratinocytes in OLP lesions and IFN-γ gene expression and protein secretion by OLP lesional T-cells in vitro (Simark-Mattsson et al., 1999; Khan et al., 2001, submitted). A previous study identified IFN-γ gene expression by lymphocytes adjacent to the epithelial basement membrane in OLP in situ (Simark-Mattsson et al., 1998). Analysis of these data together suggests that LCs or keratinocytes in OLP may present antigen associated with MHC class II to CD4+ helper T-cells that are stimulated to secrete the Th1 cytokines IL-2 and IFN-γ. Subsequently, CD8+ cytotoxic T-cells may be activated by the combination of (i) antigen associated with MHC class I on basal keratinocytes and (ii) Th1 CD4+ T-cell-derived IL-2 and IFN-γ. Activated CD8+ cytotoxic T-cells may then trigger basal keratinocyte apoptosis in OLP. Local production of IFN-γ may maintain keratinocyte MHC class II expression, thereby contributing to disease chronicity (Morhenn and Wood, 1988; Albanesi et al., 1998).
One or two antigens
Antigens that are presented by MHC class II are processed through an endosomal cellular pathway. In contrast, antigens that are presented by MHC class I are processed through a cytosolic cellular pathway. Hence, the putative antigen presented by MHC class II to CD4+ helper T-cells in OLP may differ from that presented by MHC class I to CD8+ cytotoxic T-cells. Alternatively, a single antigen may gain access to both the endosomal and cytosolic cellular pathways of antigen presentation. For example, some viruses encode proteins that are available for cytosolic processing and expression in association with MHC class I. These viral proteins are also present on the plasma membrane and therefore are available for endosomal processing and expression in association with MHC class II (Roopenian, 1992). MHC class I antigen presentation alone may result in a transient cytotoxic T-cell response, as seen in some oral viral infections and recurrent aphthous stomatitis. MHC class II antigen presentation alone may generate Th1 CD4+ T-cells. However, in the absence of MHC class I antigen presentation to CD8+ cells, the IL-2 and IFN-γ secreted by the activated Th1 CD4+ T-cells would be cytotoxically inert. As discussed below, Th1 cytokine secretion by CD4+ T-cells may be up-regulated by antigen-stimulated CD8+ T-cells, implying cross-talk between CD8+ and CD4+ T-cells in OLP. Whether one antigen or two different antigens are involved in disease pathogenesis, it is likely that simultaneous antigen presentation to CD8+ and CD4+ T-cells in the context of MHC class I and II, respectively, is required to develop persistent T-cell infiltration and CD8+ cytotoxic T-cell activity in OLP.
Th1 cytokine bias
IFN-γ and TNF-α were expressed by T-cells in the subepithelial lymphocytic infiltrate in OLP. In addition, OLP lesional T-cells contained mRNA for IFN-γ and TNF-α and secreted IFN-γ and TNF-α in vitro. OLP lesional T-cells did not secrete IL-4, IL-10, or TGF-β1 in vitro (Simark-Mattsson et al., 1998, 1999; Khan et al., 2001, submitted). We also identified elevated levels of TNF-α in the serum of OLP patients (Sugerman et al., 1996). Clearly, the basis of this CD4+ Th1 cytokine bias in OLP warrants further investigation. A role for CD40 and CD80 expression and IL-12 secretion by MHC class II+ APC should be considered, as should CD154 (CD40 receptor), CD28 (CD80 receptor), and IL-12 receptor expression, by infiltrating CD4+ T-cells in OLP (Constant and Bottomly, 1997). Th1 cytokine secretion by CD4+ T-cells in OLP may also be up-regulated by antigen-stimulated CD8+ T-cells, i.e., cross-talk between CD8+ and CD4+ T-cells may further skew the CD4+ T-cell response toward a Th1 cytokine profile in OLP.
T-cells in the lymphocytic infiltrate in OLP produce and secrete IFN-γ and TNF-α (Simark-Mattsson et al., 1998, 1999; Khan et al., 2001, submitted). As previously discussed, IFN-γ secretion by Th1 CD4+ T-cells in OLP may stimulate TNF-α secretion by CD8+ cytotoxic T-cells which triggers keratinocyte apoptosis. Blockade of IFN-γ (Debray-Sachs et al., 1991; Nicoletti et al., 1997; Prud’homme and Chang, 1999; Sigidin et al., 2001) or TNF-α (Piguet et al., 1992; Williams et al., 1992; Elliott et al., 1993, 1994) was therapeutic in human and experimental autoimmunity and may therefore be effective in the treatment of OLP. Th1 differentiation of CD4+ T-cells leading to IFN-γ secretion is stimulated by CD80 (B7-1) expression and IL-12 secretion by MHC class II+ APC (Constant and Bottomly, 1997). CD80 expression was up-regulated in multiple sclerosis (MS) plaques, while blockade of CD80 function was therapeutic in an animal model of MS (Miller et al., 1995; Windhagen et al., 1995). Recent studies detected high levels of IL-12 expression in MS plaques and rheumatoid arthritis (RA) synovia (Windhagen et al., 1995; Morita et al., 1998). IL-12 administration accelerated diabetes development and arthritis severity in animal models of insulin-dependent diabetes mellitus (IDDM) and RA (Trembleau et al., 1995; Leung et al., 2000). Reduction of IL-12 activity with neutralizing anti-IL-12 antibody or IL-12 p40 (a natural antagonist of IL-12) significantly reduced clinical disease in animal models of MS, RA, and IDDM (Leonard et al., 1995; Trembleau et al., 1997; Malfait et al., 1998; Matthys et al., 1998; Nicoletti et al., 1999; Costa et al., 2001). Hence, IL-12 may be pathogenic while IL-12 inhibition may be therapeutic in autoimmune diseases where T-cells play an important role. Although IL-12 activity in OLP has not been measured directly, the Th1 cytokine bias in OLP suggests IL-12 co-stimulation of CD4+ T-cells with subsequent production of IFN-γ. In this context, reduction of IL-12 activity may be of clinical benefit in OLP. IL-12 production by monocytes, dendritic cells, and endothelial cells is stimulated by ligation between cell-surface CD40 and T-cell CD154 (Shu et al., 1995; Cella et al., 1996; Kelsall et al., 1996; Kennedy et al., 1996; Lienenluke et al., 2000). IL-12 secretion stimulated by CD40-CD154 ligation is implicated in MS and RA (Gerritse et al., 1996; Balashov et al., 1997; MacDonald et al., 1997; Sekine et al., 1998). Disruption of CD40-CD154 interaction was preventive or therapeutic in animal models of MS, RA, and IDDM (Durie et al., 1993; Gerritse et al., 1996; Balasa et al., 1997; Boon et al., 2001). The role of CD40-CD154 ligation in OLP is unknown, although disruption of this interaction may reduce IL-12 secretion by APC and Th1 differentiation of CD4+ T-cells and may thus be therapeutic in OLP.
Hypothesis for antigen presentation and T-cell activation in OLP
The role of CD8+ T-cells is to seek out and destroy cells expressing foreign antigenic peptides in the context of MHC class I. However, the CD8+ T-cell may require antigen-specific confirmation from a CD4+ T-cell prior to initiating target cell lysis. Hence, during OLP lesion formation, the CD8+ T-cell antigen receptor may engage a specific foreign antigen (Ag 1) in the context of MHC class I expressed by basal keratinocytes [1] (numbers in brackets refer to Fig. 1). The CD8+ T-cell may then seek CD4+ T-cell confirmation by expressing the hypothetical “request cytotoxic activity” (RCA) cell-surface molecule [2] or secreting certain cytokines. The CD4+ T-cell expresses the hypothetical “RCA receptor” (RCA R) [4], but only following CD4+ T-cell antigen receptor engagement of a related foreign antigen (Ag 2) in the context of MHC class II expressed by basal keratinocytes or LCs [3]. As discussed above, Ag 1 and Ag 2 may be identical or different peptides. If different, it seems reasonable that they should have a common origin. Ligation between RCA and RCA R in combination with co-stimulatory signals from the MHC class II+ APC (e.g., CD40, CD80, and IL-12) initiates Th1 differentiation of the CD4+ T-cell that then secretes IL-2 and IFN-γ [5]. Receptors for IL-2 and IFN-γ are expressed by the CD8+ T-cell, but only following (i) specific engagement of the CD8+ T-cell antigen receptor in the context of MHC class I and/or (ii) ligation between RCA and RCA R. The CD4+ Th1 cytokines (IL-2 and IFN-γ) are detected by the CD8+ T-cell and are interpreted as confirmation to proceed with target cell (basal keratinocyte) lysis. Recent studies identified monokine induced by interferon gamma (MIG), interferon gamma-inducible protein-10 (IP-10), and macrophage chemoattractant protein-1 (MCP-1) chemokine expression by basal keratinocytes in cutaneous lichen planus lesions in situ (Spandau et al., 1998) and IL-8, MCP-1, and GRO gamma chemokine expression by IL-1-stimulated human oral keratinocytes in vitro (Bickel et al., 1996). In addition, oral keratinocytes from patients with OLP secreted cytokines that up-regulated mononuclear cell adhesion molecule expression and trans-endothelial cell migration in vitro (Yamamoto et al., 2000). Hence, keratinocyte activation by (i) the CD4+ or CD8+ T-cell following receptor-antigen-MHC trimerization or (ii) exogenous agents such as viral infection, bacterial products, mechanical trauma, systemic drugs, or contact sensitivity may up-regulate keratinocyte cytokine and chemokine secretion [6] that promotes lymphocyte extravasation and directs lymphocyte migration into the site of the developing OLP lesion.
This hypothesis predicts that both CD4+ and CD8+ T-cells must engage related foreign antigens prior to the initiation of target cell lysis. The presence of both CD4+ and CD8+ autoreactive T-cells seems unlikely. Hence, this “two-cell hypothesis” for cell-mediated cytotoxicity provides a level of protection against devastating cell-mediated autoimmune reactions. In OLP (and possibly other T-cell-mediated autoimmune diseases), this autoimmune defense mechanism may be overridden by (i) the presence of both CD4+ and CD8+ autoreactive T-cells, (ii) constitutive Th1 cytokine secretion by CD4+ lesional T-cells, (iii) Th1 cytokine secretion by the APC (as discussed above), or (iv) constitutive lytic activity of antigen-specific CD8+ lesional T-cells. The latter two hypotheses predict that CD4+ T-cells are not necessary for keratinocyte apoptosis triggered by CD8+ cytotoxic T-cells in OLP. However, the presence of large numbers of CD4+ T-cells in OLP lesions argues favorably for their involvement in disease pathogenesis.
Summary of antigen specificity in OLP
Analysis of these data suggests that many antigen-specific mechanisms may be involved in the pathogenesis of OLP, including (i) MHC class I- and MHC class II-restricted antigen presentation by lesional keratinocytes, (ii) activation of antigen-specific CD4+ helper T-cells and CD8+ cytotoxic T-cells, (iii) clonal expansion of antigen-specific T-cells, and (iv) keratinocyte apoptosis triggered by antigen-specific CD8+ cytotoxic T-cells. However, our recent studies showed that a significant proportion of OLP lesional T-cells were non-clonal (Zhou et al., 1996) and that not all CD8+ T-cell clones isolated from lichen planus lesions were cytotoxic against autologous lesional keratinocytes in vitro (Sugerman et al., 2000b). Other studies found that many intra-epithelial T-cells in OLP expressed the naïve T-cell marker CD45RA (Walton et al., 1998). Analysis of these data suggests that a proportion of T-cells in the OLP lymphocytic infiltrate are not specific for the lichen planus antigen and are not activated. As discussed below, non-specific T-cells may be attracted to and retained within OLP lesions by various mechanisms associated with pre-existing inflammation.
Non-specific Mechanisms in OLP
The epithelial basement membrane
As discussed above, epithelial basement membrane changes are common in OLP and include breaks, branches, and duplications (Jungell et al., 1989a; Zhou et al., 2001). Keratinocytes contribute to the structure of the epithelial basement membrane by secreting collagen IV and laminin V into the basement membrane zone (Marinkovich et al., 1993). Presumably, apoptotic keratinocytes are no longer able to perform this function. Hence, keratinocyte apoptosis triggered by intra-epithelial CD8+ cytotoxic T-cells may result in epithelial basement membrane disruption in OLP. Conversely, evidence from the involuting mouse mammary gland model suggests that keratinocytes require a basement-membrane-derived cell survival signal to prevent the onset of apoptosis (Pullan et al., 1996). Hence, epithelial basement membrane disruption may trigger keratinocyte apoptosis in OLP. An intriguing question in OLP is which came first — keratinocyte apoptosis or epithelial basement membrane disruption? Both mechanisms may be involved in the pathogenesis of OLP, e.g., basement membrane disruption may trigger keratinocyte apoptosis, and apoptotic keratinocytes may be unable to repair the disrupted basement membrane. Such a cyclical mechanism may underlie disease chronicity.
Matrix metalloproteinases
We recently examined the distribution, activation, and cellular sources of matrix metalloproteinases (MMPs) in OLP. MMPs are a family of zinc-containing endo-proteinases with at least 20 members. The principal function of MMPs is the proteolytic degradation of connective tissue matrix proteins. MMPs share biochemical properties but retain distinct substrate specificities. The gelatinases (e.g., MMP-2 and -9) cleave collagen IV, and the stromelysins (e.g., MMP-3 and -10) cleave collagen IV and laminin. MMP proteolysis is regulated by the action of endogenous inhibitors, including the tissue inhibitors of metalloproteinases (TIMPs), which form stable inactive enzyme-inhibitor complexes with MMPs or proMMPs. In our preliminary studies, staining for collagen IV and laminin showed epithelial basement membrane disruption in OLP (Zhou et al., 2001). MMP-2 and MMP-3 were expressed mainly in the OLP epithelium. MMP-9 was identified within the inflammatory infiltrate in the lamina propria, with occasional positive cells in the epithelium. Culture supernatants from OLP lesional T-cells contained a higher concentration of MMP-9 than those from OLP or healthy control peripheral blood T-cells. The concentration of TIMP-1, an inhibitor of MMP-9, followed a similar pattern. All T-cells contained mRNA for MMP-9 and TIMP-1. Importantly, lesional T-cell MMP-9 (but not TIMP-1) mRNA levels and protein secretion increased following stimulation with TNF-α. MMP-9 activity was confirmed by gelatin gel zymography. The in vitro activation rate of MMP-9 from OLP lesional T-cells was greater than that from peripheral blood T-cells of OLP patients and healthy control subjects, suggesting the presence of additional MMP-9 activators in the OLP lesional T-cell supernatants (Zhou et al., 2001). Hence, T-cells in OLP may be stimulated by TNF-α to secrete MMP-9. The antigen specificity of these T-cells is unknown, although non-specific T-cells may be activated in this manner, thereby amplifying the MMP-9 produced in OLP lesions. T-cell-secreted MMP-9 may disrupt the epithelial basement membrane in OLP lesions. The disrupted basement membrane in OLP no longer delivers the keratinocyte survival signal, which may trigger keratinocyte apoptosis. In addition, MMP-9-induced basement membrane disruption may facilitate the passage of antigen-specific CD8+ cytotoxic T-cells into the OLP epithelium, where they trigger further keratinocyte apoptosis. TNF-α is synthesized as a 233-amino-acid membrane-bound precursor protein which is proteolytically cleaved to yield the mature 157-amino-acid soluble cytokine. Precursor TNF-α is cleaved by TNF-alpha-converting enzyme (TACE), a membrane-bound disintegrin metalloproteinase (Gearing et al., 1994; McGeehan et al., 1994; Black et al., 1997; Moss et al., 1997; Itai et al., 2001). As previously discussed, we identified TNF-α secretion by OLP lesional T-cells and various MMPs in OLP. Hence, an additional role for MMPs in OLP may involve the release of active TNF-α from OLP lesional T-cells.
Mast cells
Our recent studies showed increased mast cell density in OLP (Zhao et al., 1997). Approximately 60% of mast cells were degranulated in OLP, compared with 20% in normal buccal mucosa (Zhao et al., 2001). Mast cell degranulation in OLP releases a range of pro-inflammatory mediators such as TNF-α, chymase, and tryptase. TNF-α may up-regulate endothelial cell adhesion molecule (CD62E, CD54, and CD106) expression in OLP that is required for lymphocyte adhesion to the luminal surfaces of blood vessels and subsequent extravasation (Klein et al., 1989; Walsh et al., 1991; Walton et al., 1994). In addition, we identified clusters of mast cells and intra-epithelial CD8+ T-cells at sites of basement membrane disruption in OLP (Zhou et al., 2002). Analysis of these data suggests that mast cells may play a role in epithelial basement membrane disruption in OLP, and that CD8+ T-cells may migrate through basement membrane breaks to enter the OLP epithelium. MMPs are secreted as inactive proenzymes and are rapidly degraded after activation. Chymase, a mast cell protease, is a known activator of MMP-9 (Fang et al., 1997). Hence, basement membrane disruption in OLP may be mediated by mast cell proteases directly or indirectly via activation of T-cell-secreted MMP-9 (Zhao et al., 2001; Zhou et al., 2001).
Chemokines
The chemokines are a superfamily of pro-inflammatory cytokines that are produced by virtually all somatic cells. RANTES (regulated on activation, normal T-cell expressed and secreted) is one of the most extensively studied chemokines. RANTES is a member of the CC chemokine family and is produced by various cells, including activated T-lymphocytes, bronchial epithelial cells, rheumatoid synovial fibroblasts, oral keratinocytes, and mast cells. RANTES plays a critical role in the recruitment of lymphocytes, monocytes, natural killer cells, eosinophils, basophils, and mast cells. Chemokines mediate their biological effects by binding to cell-surface receptors. Several RANTES receptors have been identified, including CCR1, CCR3, CCR4, CCR5, CCR9, and CCR10. The CC chemokines, including RANTES, activate mast cell migration and degranulation via these G protein-coupled receptors (Bischoff et al., 1993).
We identified T-cell RANTES expression in OLP in situ (Zhao et al., 2002). OLP lesional T-cells expressed mRNA for RANTES, and TNF-α stimulation up-regulated OLP lesional T-cell RANTES secretion in vitro (Zhao et al., 2001). Mast cells expressed the CCR1 RANTES receptor in OLP in situ (Zhao et al., 2002). An unidentified factor in OLP lesional T-cell supernatant up-regulated human mast cell line (HMC-1) CCR1 mRNA expression in vitro (Zhao et al., 2002). OLP lesional T-cell supernatant stimulated HMC-1 migration in vitro. This effect was partially blocked by anti-RANTES antibody (Zhao et al., 2002). OLP lesional T-cell supernatant stimulated HMC-1 degranulation in vitro with release of TNF-α and histamine. This effect was blocked by anti-RANTES antibody (Zhao et al., 2001). Hence, RANTES secreted by OLP lesional T-cells may attract mast cells into the developing OLP lesion and subsequently stimulate mast cell degranulation. Degranulating mast cells in OLP would release TNF-α, which up-regulates OLP lesional T-cell RANTES secretion. Such a cyclical mechanism may underlie OLP chronicity. Furthermore, RANTES induces expression of PI 3-kinase, which is involved in signal transduction for both chemotaxis and mitogen-activated protein kinase activation. PI 3-kinase activates Akt/protein kinase B, an important component of the cell’s pro-survival machinery (Kane et al., 1999). We identified CCR1 expression by both T-cells and mast cells in OLP (Zhao et al., 2002). Hence, in addition to stimulating mast cell chemotaxis and degranulation, RANTES secreted by OLP lesional T-cells may also prolong the survival of inflammatory cells in OLP and thereby contribute to disease chronicity. As discussed, a previous study identified basal keratinocyte expression of another CC chemokine, MCP-1, and two CXC chemokines, MIG and IP-10, in cutaneous lichen planus lesions (Spandau et al., 1998). Analysis of these data suggests that chemokines produced by T-cells and keratinocytes may attract various inflammatory cells into the site of the developing OLP lesion. The antigen specificity of the T-cells recruited in this manner is unknown. However, constitutive chemokine receptor expression on naïve T-cells (Sallusto et al., 1998) suggests that such a mechanism may result in the accumulation of non-specific T-cells in OLP lesions.
Summary of non-specific mechanisms in OLP
Analysis of these data suggests that many non-specific mechanisms may be involved in the pathogenesis of OLP, including (i) mast cell chemotaxis and degranulation stimulated by T-cell RANTES, (ii) endothelial cell adhesion molecule expression stimulated by mast cell TNF-α, (iii) T-cell MMP-9 activation by mast cell chymase, (iv) epithelial basement membrane disruption by mast cell proteases or T-cell MMP-9, (v) keratinocyte apoptosis triggered by epithelial basement membrane disruption, (vi) intra-epithelial CD8+ T-cell migration through basement membrane breaks, (vii) inflammatory cell survival prolonged by T-cell RANTES, and (viii) non-specific T-cell recruitment by keratinocyte-derived chemokines. We suggested previously that only a small percentage of lymphocytes recruited to the OLP lesion site are specific for the lichen planus antigen (Sugerman et al., 2000a). Non-specific T-cells in OLP may contribute to disease pathogenesis by secreting RANTES and MMP-9, although this remains to be determined.
Deficient Antigen-specific Immunosuppression in OLP
In our recent studies, the expression of TGF-β1 in the subepithelial lymphocytic infiltrate in OLP was variable, with regions of positive and negative expression. The intra-epithelial lymphocytes in OLP were all TGF-β1- (Khan et al., 2001, submitted). TGF-β1 inhibits growth and induces differentiation and apoptosis of keratinocytes in vitro (Flanders and Roberts, 2001). Hence, T-cell-derived TGF-β1 may play a role in the epithelial pathology associated with OLP. TGF-β-secreting T-cells were recognized recently as a distinct population of antigen-specific CD4+ regulatory T-cells, termed “Th3” (Fukaura et al., 1996). There is now compelling evidence that antigen-specific CD4+ TGF-β-secreting Th3 regulatory T-cells suppress immune responses to self-antigens and prevent autoimmunity (Bridoux et al., 1997; Mason and Powrie, 1998). TGF-β1 exerts its immunosuppressive effect in part by interfering with antigen presentation, thereby suppressing effector T-cell proliferation, differentiation, and cytokine secretion. TGF-β1 down-regulates APC IL-12 production, thus blocking Th1 differentiation of CD4+ T-cells, IFN-γ secretion, and cytotoxic T-cell responses (Letterio and Roberts, 1998). TGF-β1 activity is mediated via the TGF-βII receptor, with subsequent phosphorylation of the TGF-βI receptor. Both receptors form a heterodimeric complex that initiates TGF-β1 activity (Massagué, 1998). The role of the TGF-βIII receptor in TGF-β signaling is currently under investigation (Blobe et al., 2001). The activated TGF-βI receptor phosphorylates cytoplasmic Smad2 or Smad3, which then binds Smad4. The Smad complex translocates to the nucleus, where it regulates the transcription of TGF-β response genes. Of interest in the current context is that mice deficient in TGF-β1 or Smad3 developed widespread autoimmune-like inflammatory disease, including a periductal lymphocytic infiltrate in the liver, pancreas, and salivary glands (Shull et al., 1992; Dang et al., 1995; Yang et al., 1999). Exogenously administered TGF-β1 was both preventive and therapeutic in animal models of autoimmune disease, including experimental allergic encephalomyelitis and collagen-induced arthritis (Johns et al., 1991; Kuruvilla et al., 1991; Racke et al., 1991; Thorbecke et al., 1992). Retroviral transduction of T-cells with TGF-β1 cDNA significantly delayed the onset of experimental allergic encephalomyelitis. The transduced T-cells secreted significant quantities of TGF-β1, and the clinical benefit was negated by simultaneous injection of anti-TGF-β1 antibody (Chen et al., 1998). Finally, administration of neutralizing anti-TGF-β1 antibodies exacerbated experimental allergic encephalomyelitis, suggesting that endogenous TGF-β1 has a regulatory function in this animal model of MS (Racke et al., 1992; Johns and Sriram, 1993). Analysis of these data suggests that TGF-β1 deficiency may predispose to autoimmune lymphocytic inflammation, while TGF-β1 administration may be therapeutic in autoimmune diseases where T-cells play an important role.
In this context, OLP chronicity may be due, in part, to a defect in the TGF-β1 immunosuppressive pathway involving (i) insufficient numbers of TGF-β1-secreting Th3 regulatory T-cells, (ii) blockage of TGF-β1 secretion, (iii) secretion of non-functional TGF-β1, (iv) defective or inadequate TGF-β1 receptor expression, or (v) defective intracellular signaling downstream from the TGF-β1 receptors. In our recent studies, the intra-epithelial lymphocytes in OLP were all TGF-β1-, suggesting insufficient numbers of regulatory T-cells in the OLP epithelium. We identified TGF-β1+ T-cells in the subepithelial lymphocytic infiltrate in OLP, although OLP lesional T-cells did not secrete TGF-β1 in vitro, suggesting that T-cell TGF-β1 secretion may be blocked in OLP (Khan et al., 2001, submitted). Importantly, the Th1 cytokine IFN-γ inhibits the immunosuppressive activity of TGF-β1 by blocking TGF-β1-induced phosphorylation of the Smad3 transcription factor (Ulloa et al., 1999). Hence, the balance between TGF-β1 and IFN-γ signaling may determine the level of immunological activity in OLP lesions, as has been suggested in other inflammatory mucosal diseases (Strober et al., 1997). Local overproduction of IFN-γ by Th1 CD4+ T-cells in OLP lesions would down-regulate the immunosuppressive effect of TGF-β1 and up-regulate keratinocyte MHC class II expression and CD8+ cytotoxic T-cell activity.
Breakdown of Immune Privilege in OLP
In recent studies, TNF-α was expressed as a continuous band by basal epithelial cells, and TNF R1 was expressed by infiltrating T-cells in OLP (Khan et al., 2001, submitted). Analysis of these data suggests that, in OLP, keratinocyte-derived TNF-α may trigger T-cell apoptosis via TNF R1. Hence, disease activity in OLP may be determined by the balance between keratinocyte apoptosis triggered by infiltrating T-cells and T-cell apoptosis triggered by resident keratinocytes. In this context, OLP may result from a failure of resident keratinocytes to trigger T-cell apoptosis. Oral keratinocytes in OLP may fail to express enough active TNF-α, or the release of TNF-α from the surfaces of oral keratinocytes in OLP may be blocked, possibly due to defective MMP activity (above). Infiltrating T-cells in OLP may fail to express enough active TNF R1, or the apoptotic pathway downstream of TNF R1 (including TRADD, FADD, and caspases 8, 1, and 3) may be defective in OLP lesional T-cells. The concept of inadequate keratinocyte-derived TNF-α-mediated T-cell lysis in OLP is purely speculative, although supported indirectly by findings of keratinocyte TNF-α expression in the normal oral mucosa (Walsh et al., 1995) and skin (Kristensen et al., 1993). In more general terms, the normal oral mucosa may be an immune-privileged site, similar to the eye, testis, and placenta. In the eye and testis, immune privilege is mediated by Fas ligand (CD95L), expressed by stromal cells, that triggers apoptosis of infiltrating inflammatory cells expressing Fas (CD95) (Bellgrau et al., 1995; Griffith et al., 1995). This mechanism is thought to minimize potentially damaging inflammation in these organs. A similar mechanism (oral keratinocyte CD95L or TNF-α triggering T-cell apoptosis via CD95 or TNF R1, respectively) may prevent excessive T-cell infiltration in the normal oral mucosa, while failure of such a mechanism may result in OLP and possibly other autoimmune oral mucosal diseases, including cicatricial pemphigoid and pemphigus vulgaris. This concept has broad-reaching implications for many T-cell-mediated autoimmune diseases. MS, IDDM, Sjögren’s syndrome (SS), and OLP may result from: (i) inadequate CD95L or TNF-α expression by neural Schwann cells, pancreatic islet cells, salivary acinar cells, and oral keratinocytes (respectively); (ii) inadequate CD95 or TNF R1 expression by infiltrating T-cells; or (iii) defective T-cell apoptotic pathways downstream from CD95 or TNF R1. A role for CD95 in the prevention of autoimmunity was suggested by the CD95 (lpr) and CD95L (gld) mutations that are associated with spontaneous lymphoproliferative autoimmune diseases in mice (Chu et al., 1993; Takahashi et al., 1994). Such a role for TNF-α in the prevention of autoimmunity has not been identified.
An alternative explanation for these findings is that keratinocyte-derived TNF-α may trigger T-cell apoptosis via TNF R1 in OLP, although killing may be restricted to immunosuppressive T-cells such as those producing TGF-β1. As discussed above, we showed recently that intra-epithelial T-cells in OLP did not express the immunosuppressive cytokine TGF-β1, suggesting unchecked immunological activity within the OLP lesional epithelium (Khan et al., 2001, submitted). Selective lysis of intra-epithelial TNF R1+ TGF-β1+ immunosuppressive T-cells by keratinocyte-derived TNF-α may promote disease activity in OLP. This hypothesis suggests that the apoptotic pathway (TNF R1, TRADD, FADD, and caspases 8, 1, and 3) is fully functional in immunosuppressive T-cells yet non-functional in cytotoxic T-cells in OLP. Furthermore, many T-cell-mediated autoimmune diseases (including MS, IDDM, SS, and OLP) may be associated with up-regulated TNF R1 or CD95 expression and apoptosis of TGF-β1+ immunosuppressive T-cells, resulting in unchecked immunological activity at the autoimmune lesion site. We also identified weak keratinocyte TGF-β1 expression in OLP (Khan et al., 2001, submitted). TGF-β1 is expressed in normal adult mouse epidermis and papillary dermis and may play a role in suppressing unwanted immune response in the oral mucosa and skin (Thompson et al., 1989). TGF-β1 is also secreted by a variety of tumors and may be involved in the suppression of anti-tumor immune responses (Gorelik and Flavell, 2001; Pasche, 2001). In this context, keratinocyte-derived TGF-β1-mediated immunosuppression may be defective in OLP lesions, although further studies are required to address this question.
Graft-vs.-Host Disease and OLP
Graft-vs.-host disease (GVHD) is a common serious complication following allogeneic bone marrow transplantation (BMT). Most patients who undergo allogeneic BMT receive stem cells from MHC-identical donors. In these patients, GVHD is initiated by donor T-cells that recognize a subset of host peptides called minor histocompatibility antigens (miHAs). miHAs are derived from the expression of polymorphic genes that distinguish host from donor. Recent studies have shown that host-derived, rather than donor-derived, APC initiate GVHD (Shlomchik et al., 1999). Acute GVHD occurs within the first 100 days of transplantation and consists of the triad of dermatitis, enteritis, and hepatitis, with immunosuppression and cachexia. Acute cutaneous GVHD usually starts as scattered erythematous macules and papules. Erythematous macules may coalesce to form confluent erythema. Chronic GVHD develops after day 100 and consists of an autoimmune syndrome affecting multiple organs. The skin is the main organ involved in chronic GVHD and manifests as a lichen-planus-like eruption or scleroderma (Kuechle, 2001).
Oral involvement occurs in 33% to 75% of patients with acute GVHD and up to 80% of patients with chronic GVHD (Schubert and Sullivan, 1990). Oral mucosal GVHD resembles OLP both clinically and histologically (Fujii et al., 1988; Mattsson et al., 1992). As with OLP, SCC may develop in oral and cutaneous chronic GVHD (Otsubo et al., 1997; Gmeinhart et al., 1999). Although the antigen specificity of lichen planus and mucocutaneous GVHD is probably distinct, it is likely that they share similar immunological effector mechanisms, resulting in T-cell infiltration, basal keratinocyte apoptosis, epithelial basement membrane disruption, and clinical disease. Hence, research findings in one disease may give clues to the pathophysiology of the other. The role of TNF-α as a major effector molecule of GVHD has been confirmed in several experimental systems. Importantly, neutralizing anti-TNF-α antibodies have been shown to alleviate cutaneous and intestinal GVHD in both mice and humans (Piguet et al., 1987; Herve et al., 1992; Brown et al., 1999; Stuber et al., 1999). Blockade of the CD40-CD154 co-stimulatory pathway of antigen presentation prevented GVHD following allogeneic BMT (Durie et al., 1994; Blazar et al., 1998; Seung et al., 2000). As discussed previously, CD40-CD154 blockade inhibits APC IL-12 production and Th1 differentiation of CD4+ T-cells, thereby suppressing cell-mediated immune responses and GVHD. The role of the Fas apoptotic pathway in cutaneous GVHD is less clear. In one study, the transfer of cells lacking Fas-L (CD95L) reduced the severity of murine cutaneous GVHD (Miwa et al., 1999). In another study, recipient mice deficient in Fas (CD95) showed increased severity of cutaneous GVHD (van den Brink et al., 2000). An MMP inhibitor was recently shown to alleviate GVHD pathology in the liver, intestine, and hematopoietic tissues and to reduce weight loss and mortality in murine GVHD (Hattori et al., 1999). Mast cells may undergo extensive degranulation during the development of murine cutaneous GVHD (Claman, 1985). Mast cell degranulation and epithelial cell damage preceded the influx of effector lymphocytes in murine GVHD (Murphy et al., 1994). In another study, mast-cell-deficient mice developed intestinal GVHD that was indistinguishable from that in their mast-cell-competent littermates (Newlands et al., 1990). Hence, the role of mast cells in GVHD is unclear. Current studies in a murine model may elucidate the genetic and cellular pathogenesis of mucocutaneous GVHD.
Keratinocyte Apoptosis and LC Maturation in OLP
Not all auto-reactive T-cells are deleted in the thymus (Gammon and Sercarz, 1989). Why, then, do some individuals carrying auto-reactive T-cells develop autoimmune disease while others do not? The outcome of self-antigen presentation depends on the activation state of the APC. To stimulate a T-cell response, dendritic cells (DCs) and presumably LCs must undergo a process of terminal differentiation called “maturation”. Stimuli for DC and LC maturation include inflammatory cytokines (IL-1β, TNF-α), CD40L (CD154) expressed by activated T-cells, necrotic cells, HSPs, nucleotides, reactive oxygen intermediates, neurotransmitters, MMP-9, extracellular matrix degradation products, mechanical trauma, various allergens, ion channel blockade, Fc receptor aggregation, viral RNA, and bacterial lipopolysaccharide (Steinman et al., 2000; Gallucci and Matzinger, 2001). DCs and LCs endocytose apoptotic cells including keratinocytes and migrate to the regional lymph nodes, where they present peptides derived from the apoptotic cells on MHC classes I and II (Albert et al., 1998a,b; Inaba et al., 1998; Rovere et al., 1998; Huang et al., 2000). Under normal circumstances, APCs carrying self-peptides derived from apoptotic cells do not receive a maturation stimulus and therefore do not trigger an auto-reactive T-cell response (Steinman et al., 2000; Gallucci and Matzinger, 2001). Immature APCs may avoid activating self-reactive T-cells by various means, including (i) failure to form MHC-peptide complexes, (ii) absence of co-stimulatory molecule expression, or (iii) direct killing of self-reactive T-cells (Suss and Shortman, 1996; Steinman et al., 2000). Conversely, APC endocytosis of apoptotic cells followed by APC maturation may activate self-reactive CD4+ T-cells that differentiate into Th1 or Th2 phenotypes and promote cell- or antibody-mediated autoimmune reactions. The nature of the APC maturation stimulus (e.g., cytokines, CD40L, necrotic cells, HSPs, etc.) may determine the outcome (Th1 vs. Th2) of CD4+ T-cell activation.
Endocytosis of apoptotic vaginal keratinocytes by LCs during the estrus cycle has been described in mice (Parr et al., 1991). Physiological keratinocyte apoptosis in the normal oral mucosa is associated with normal epithelial turnover (Birchall et al., 1995). Oral LCs may endocytose apoptotic oral keratinocytes and migrate to the regional lymph nodes. In the normal oral mucosa, the LCs do not receive a maturation stimulus. Hence, physiological oral keratinocyte apoptosis does not elicit an anti-keratinocyte autoimmune T-cell response, even in the presence of keratinocyte-specific T-cells. As discussed previously, OLP and oral lichenoid lesions may be triggered or exacerbated by mechanical trauma, viral infection, bacterial products, systemic drugs, or contact allergens which up-regulate oral keratinocyte HSP expression and stimulate an anti-HSP autoimmune T-cell response (Sugerman et al., 1995). An alternative explanation is that these exogenous agents and/or the up-regulated HSP itself may stimulate oral LC maturation (Steinman et al., 2000; Gallucci and Matzinger, 2001). In the regional lymph node, mature LCs that have endocytosed apoptotic oral keratinocytes would then activate keratinocyte-specific CD4+ T-cells. As discussed previously, the outcome (Th1 vs. Th2) of CD4+ T-cell activation may depend on the nature of the maturation stimulus delivered to the LC. The Th1 or Th2 cytokines released from the activated keratinocyte-specific CD4+ helper T-cells would co-stimulate keratinocyte-specific CD8+ T-cells or B-cells, resulting in local or widespread cell- or antibody-mediated anti-keratinocyte autoimmune inflammation. The phenotype of the resultant oral mucosal pathology would depend on the precise keratinocyte peptide presented by the mature LC and the cytokine profile (Th1 or Th2) of the responding CD4+ helper T-cell. In this context, a basal keratinocyte peptide triggering a Th1 response may activate anti-keratinocyte auto-reactive CD8+ cytotoxic T-cells, resulting in OLP. A keratinocyte desmosomal peptide (desmoglein-3) triggering a Th2 response may stimulate auto-reactive B-cell anti-desmoglein-3 antibody production, resulting in pemphigus vulgaris. A keratinocyte hemidesmosomal (BP-180) or basement membrane (laminin 5) peptide triggering a Th2 response may stimulate auto-reactive B-cell anti-BP-180 or anti-laminin 5 antibody production, resulting in cicatricial pemphigoid.
As discussed previously, a microbe or systemic drug may provide the LC maturation signal. In this case, the activation of anti-keratinocyte auto-reactive lymphocytes would cease upon microbe clearance or drug withdrawal. Erythema multiforme triggered by herpes simplex virus or a systemic drug is an example of this type of inflammation in the oral cavity. Long-term autoimmune oral mucosal pathology (e.g., OLP, pemphigus vulgaris, cicatricial pemphigoid) may result from (i) failure to eliminate the LC maturation stimulus (e.g., microbe, rough restoration, sharp tooth, systemic drug), (ii) persistence of mature LCs, or (iii) failure to suppress autoreactive lymphocytes in the oral mucosa. Furthermore, once OLP has been initiated, the combination of cell-mediated cytotoxicity and various inflammatory cytokines would continue to provide both the apoptotic oral keratinocytes and the oral LC maturation stimulus, thus contributing to lesion chronicity. Little is known about the role of oral LCs and the regional lymphatics in OLP lesion formation or chronicity. However, blockade of oral LC maturation or migration to the regional lymph nodes may prevent anti-keratinocyte auto-reactive T-cell activation and may thus be therapeutic in OLP.
In the case of oral mucosal GVHD, cytotoxic drugs or radiation may provide both the apoptotic oral keratinocytes and the oral LC maturation stimulus resulting in chronic oral lichenoid lesions following allogeneic BMT. Although Grinspan’s syndrome (triad of OLP, IDDM, and hypertension) has been described, little evidence supports a connection between IDDM and OLP. The oral lichenoid lesions in Grinspan’s syndrome may be triggered by the drugs used to treat IDDM or hypertension (Scully et al., 1998). However, recent studies have suggested that hyperglycemia-induced apoptosis may underlie the islet β-cell loss (Donath et al., 1999), congenital malformations (Moley et al., 1998), microangiopathy (Ho et al., 2000), neuropathy (Delaney et al., 2001), retinopathy (Naruse et al., 2000), cardiomyopathy (Dyntar et al., 2001), and nephropathy (Ortiz et al., 1997) associated with IDDM. It is tempting to speculate that hyperglycemia may also trigger oral keratinocyte apoptosis and stimulate oral LC maturation, thereby causing the oral lichenoid lesions associated occasionally with IDDM. Similarly, paraneoplastic pemphigus, pemphigoid, and lichenoid lesions may result from tumor cell apoptosis and APC maturation (possibly stimulated by tumor-associated HSP) with subsequent antibody- and cell-mediated mucocutaneous autoimmune inflammation (Setterfield et al., 1999; Allen and Camisa, 2000; Bowen et al., 2000; Sauter et al., 2000).
Carcinogenesis in OLP
SCC arises occasionally at the site of a pre-existing OLP lesion, although it is unlikely that OLP is inherently pre-malignant (Eisenberg, 2000; Silverman, 2000). The cause of increased oral cancer risk in OLP patients is unknown, although the oral mucosa affected by OLP may be more sensitive to exogenous mutagens in tobacco, alcohol, betel quid, and Candida albicans. As discussed previously, some cases of malignant transformation of OLP lesions may represent an initial lichenoid response to pre-existing tumor cells expressing elevated levels of HSP, followed by the development of overt clinical and histological malignancy. We recently identified TGF-β1 expression in the subepithelial lymphocytic infiltrate in OLP (Khan et al., 2001, submitted). T-cell-derived TGF-β1 may inhibit growth and induce differentiation and apoptosis of oral mucosal keratinocytes, thereby suppressing tumor formation (Flanders and Roberts, 2001). T-cells in OLP also express IFN-γ and TNF-α (Simark-Mattsson et al., 1998, 1999; Khan et al., 2001, submitted). while many studies have shown that TNF-α, IFN-γ, and IL-12 inhibit tumor growth and metastasis (Aggarwal et al., 2001; Billiau and Vanderbroeck, 2001; Esche et al., 2001). Hence, TGF-β1, TNF-α, IFN-γ, and IL-12 may inhibit carcinogenesis in OLP.
As discussed previously, TGF-β1 was expressed weakly by keratinocytes in OLP (Khan et al., 2001, submitted). In previous studies, mouse skin treated with the tumor promoter 12-tetradecanoyl-phorbol-13-acetate showed high levels of TGF-β1 mRNA in the suprabasal epithelium (Akhurst et al., 1988). Overexpression of TGF-β1 in mouse skin resulted in stage-dependent protection from, or enhancement of, carcinogen-induced tumorigenesis (Cui et al., 1996). In this context, keratinocyte TGF-β1 expression may play a role in cancer development in OLP. The chronic inflammatory response and simultaneous epithelial wound-healing response may increase the likelihood of cancer-forming gene mutations in OLP. This hypothesis is supported by recent important findings linking chemical mediators of T-cell inflammation to tumorigenesis. These studies showed that macrophage migration inhibitory factor (MIF) stimulated tumor cell proliferation, while MIF neutralization inhibited tumor growth (Takahashi et al., 1998; Chesney et al., 1999; Shimizu et al., 1999). Importantly, MIF released from T-cells and macrophages suppressed the transcriptional activity of the p53 tumor suppressor protein (Hudson et al., 1999). As discussed previously, normal p53 function is central to the prevention of many cancers, including oral SCC (Sugerman and Savage, 1999). Blockade of keratinocyte p53 function by MIF may allow growth-promoting gene mutations to go unchecked, thus setting the stage for cancer development in OLP. MIF plays an essential role in delayed-type hypersensitivity reactions, although its role in OLP has not been reported (Bernhagen et al., 1996). Our previous studies identified MMP-9 production by T-cells in OLP (Zhou et al., 2001). MMP-9 derived from mast cells, neutrophils, and macrophages promoted cutaneous carcinogenesis in a K14-HPV16 transgenic mouse model via paracrine effects on oncogene-positive neoplastic cells (Coussens et al., 2000). Hence, MIF, MMP-9, and keratinocyte TGF-β1 may promote carcinogenesis in OLP.
In summary, the integrated signal from various tumor inhibitors (e.g., TGF-β1, TNF-α, IFN-γ, IL-12) and promoters (e.g., MIF, MMP-9, keratinocyte TGF-β1) may determine the sensitivity of oral keratinocytes to exogenous mutagens and may regulate tumor growth and metastasis following cancer formation in OLP. Of further interest, the level of TGF-β1 in OLP may be pivotal in disease pathogenesis and outcomes. Inadequate immunosuppression associated with low TGF-β1 activity may promote hyperactive immune responses in OLP. In contrast, high TGF-β1 activity may suppress antitumor immune responses and thus promote carcinogenesis in OLP. In more general terms, endogenous and therapeutic immunosuppression may, at first glance, appear beneficial in OLP. However, such immunosuppression may down-regulate antitumor immune responses and increase the oral cancer risk in OLP patients. It is interesting to note that SCC is more likely to develop in atrophic and erosive OLP lesions that typically receive immunosuppressive treatment. Clearly, much research is required to determine the total impact of current immunosuppressive therapies in OLP. Future therapies may down-regulate the hyperactive immune responses but maintain the antitumor immune responses in OLP.
A Unifying Hypothesis for the Pathogenesis of OLP
On the basis of our recent experimental findings, we propose a unifying hypothesis implicating both antigen-specific and non-specific mechanisms in the pathogenesis of OLP. A lichen planus antigen is expressed in association with MHC class I molecules on keratinocytes at the OLP lesion site [1] (numbers in brackets refer to Fig. 2). Antigen-specific CD8+ cytotoxic T-lymphocytes (CTLs) are activated in the OLP epithelium (possibly with help from Th1 CD4+ T-cells, as discussed previously) and trigger keratinocyte apoptosis via secreted TNF-α [2], although roles for granzyme B and Fas cannot be excluded at this stage. TNF-α may be activated and released from the CTL surface by lesional MMPs. Activated T-cells undergo intra-lesional clonal expansion and release RANTES and other cytokines [3] that up-regulate mast cell CCR1 expression and stimulate intra-lesional mast cell migration and degranulation [4]. Degranulating mast cells release TNF-α which up-regulates endothelial cell adhesion molecule expression for lymphocyte adhesion and extravasation [5]. Mast cell TNF-α also up-regulates RANTES [6] and MMP-9 [7] secretion by OLP lesional T-cells. Activated lesional T-cells (and possibly keratinocytes) secrete chemokines which attract extravasated lymphocytes toward the OLP epithelium [8]. Degranulating mast cells release chymase that damages the epithelial basement membrane directly [9] or indirectly via activation of MMP-9 secreted by OLP lesional T-cells [10]. Epithelial basement membrane disruption facilitates the passage of lymphocytes into the OLP epithelium [11] and denies keratinocytes a cell survival signal, resulting in further keratinocyte apoptosis [12].
Initial Events in OLP Lesion Formation
As discussed previously, keratinocyte antigen expression may be the initial event in OLP lesion formation. Subsequently, the lichen planus antigen is recognized by antigen-specific T-cells circulating through the oral epithelium or T-cells attracted to the oral epithelium by keratinocyte-derived chemokines. Following antigen recognition and activation, T-cell CD40L may stimulate oral LC maturation for subsequent antigen presentation. Keratinocyte antigen expression and chemokine secretion in OLP may be stimulated by viral infection, bacterial products, mechanical trauma, systemic drugs or contact sensitivity. In this scenario, mast cell degranulation and other non-specific mechanisms are downstream events in OLP lesion formation. Conversely, mast cell degranulation may be an initial event that disrupts the epithelial basement membrane and stimulates oral keratinocyte antigen expression and LC maturation. In this scenario, antigen-specific interactions between keratinocytes and T-cells are downstream events in OLP lesion formation. It is also possible that the initial event in OLP lesion formation may vary from patient to patient. However, following the initial event in a susceptible patient, both antigen-specific and non-specific mechanisms continue relentlessly, producing the chronic lesions recognized clinically and histologically as OLP. As is evident from this discussion, the initial event in OLP lesion formation is unknown. Current time-course studies in a murine model may elucidate primary events and genetic regulation in OLP.
OLP Susceptibility
It is likely that some of the antigen-specific and non-specific mechanisms described here for OLP occur together or in isolation in many people without causing chronic lichenoid lesions. For example, viral infection of the oral mucosa produces a T-cell response that typically does not progress to OLP. Similarly, mechanical trauma to the oral mucosa causes mast cell degranulation that typically does not progress to OLP. So, what constitutes OLP susceptibility? Since generalized atopy is not associated with OLP, it is unlikely that dysregulated mast cell degranulation is central to OLP susceptibility. However, dysregulated mast cell chymase or TNF-α secretion (independent of mast cell degranulation) may be involved. In this context, OLP susceptibility may result from a combination of factors, including dysregulated oral keratinocyte antigen expression, persistence of mature oral LCs, circulating auto-reactive T-cells, and defective immune suppressor activity following self-antigen recognition.
OLP Research Directions and Future Therapies
Many questions remain concerning the etiology and pathogenesis of OLP. What is the lichen planus antigen? Does the lichen planus antigen vary from site to site or patient to patient? What proportion of T-cells in OLP lesions are specific for the lichen planus antigen? What is the initial event in OLP lesion formation? What roles do oral LCs and the regional lymphatics play in OLP lesion formation and chronicity? What triggers keratinocyte apoptosis in OLP? What constitutes OLP susceptibility? Answers to these questions may help produce a cure for OLP. In the meantime, analysis of current data suggests that blocking IL-12, IFN-γ, TNF-α, RANTES, or MMP-9 activity or up-regulating TGF-β1 activity in OLP may be therapeutic.
Conclusions
The pathogenesis of OLP may involve both antigen-specific and non-specific mechanisms. Antigen-specific mechanisms in OLP include antigen presentation by basal keratinocytes and antigen-specific keratinocyte killing by CD8+ cytotoxic T-cells. Non-specific mechanisms include mast cell degranulation and matrix metalloproteinase activation in OLP lesions. The initial event in OLP lesion formation and the factors that determine OLP susceptibility are unknown. Clearly, more work is required for a full understanding of the etiology and pathogenesis of OLP.
Hypothesis for antigen presentation and T-cell activation in OLP. Initially, the CD8+ T-cell antigen receptor engages a specific foreign antigen (Ag 1) in the context of MHC class I on the basal keratinocyte target cell in OLP [1]. The CD8+ T-cell may then seek CD4+ T-cell confirmation by expressing the hypothetical “request cytotoxic activity” (RCA) cell surface molecule [2]. The CD4+ T-cell expresses the hypothetical “RCA receptor” (RCA R) [4], but only following CD4+ T-cell antigen receptor engagement of a related foreign antigen (Ag 2) in the context of MHC class II on the antigen-presenting cell (basal keratinocyte or Langerhans cell in OLP) [3]. Ligation between RCA and RCA R in combination with co-stimulatory signals from the MHC class II+ antigen-presenting cell (e.g., CD40, CD80, and IL-12 binding CD154, CD28, and IL-12 R, respectively, on the CD4+ T-cell) initiates Th1 differentiation of the CD4+ T-cell that then secretes IL-2 and IFN-γ [5]. Receptors for IL-2 and IFN-γ are expressed by the CD8+ T-cell, but only following (i) specific engagement of the CD8+ T-cell antigen receptor in the context of MHC class I and/or (ii) ligation between RCA and RCA R. The CD4+ Th1 cytokines (IL-2 and IFN-γ) are detected by the CD8+ T-cell and interpreted as confirmation to proceed with target cell (basal keratinocyte) lysis. Keratinocyte activation by (i) the CD4+ or CD8+ T-cell following receptor-antigen-MHC trimerization or (ii) exogenous agents such as viral infection, bacterial products, mechanical trauma, systemic drugs, or contact sensitivity up-regulates keratinocyte cytokine and chemokine secretion [6] that promotes lymphocyte extravasation and directs lymphocyte migration into the site of the developing OLP lesion. Unifying hypothesis for the pathogenesis of OLP. 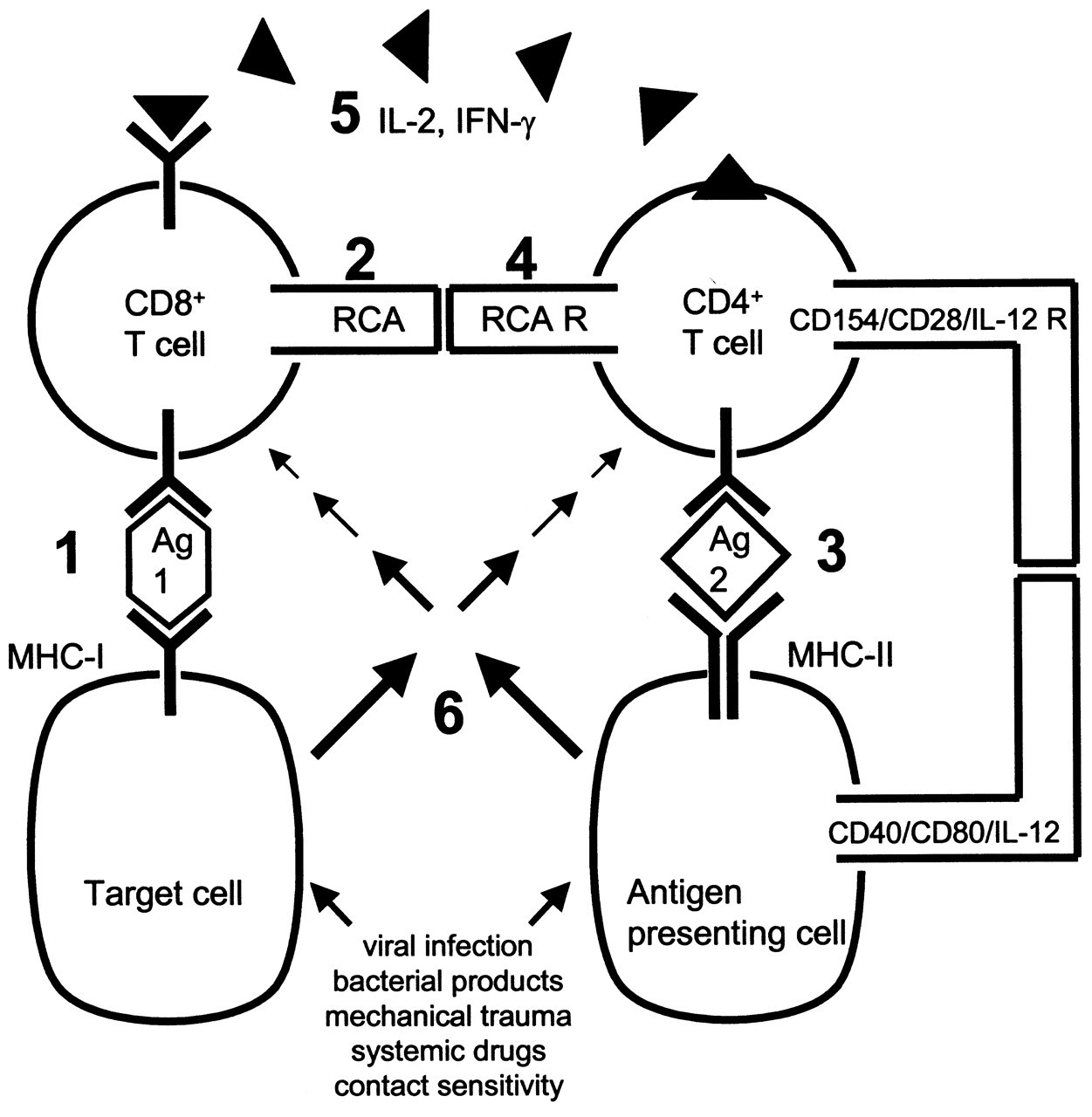
Footnotes
Acknowledgements
Philip Sugerman is supported by a National Health and Medical Research Council (Australia) Industry Research Fellowship (# 143125). This study was supported by the Australian Dental Research Foundation, Inc.