
Abstract
It is now 35 years since Brandtzaeg and Kraus (1965) published their seminal work entitled "Autoimmunity and periodontal disease". Initially, this work led to the concept that destructive periodontitis was a localized hypersensitivity reaction involving immune complex formation within the tissues. In 1970, Ivanyi and Lehner highlighted a possible role for cell-mediated immunity, which stimulated a flurry of activity centered on the role of lymphokines such as osteoclast-activating factor (OAF), macrophage-activating factor (MAF), macrophage migration inhibition factor (MIF), and myriad others. In the late 1970s and early 1980s, attention focused on the role of polymorphonuclear neutrophils, and it was thought that periodontal destruction occurred as a series of acute exacerbations. As well, at this stage doubt was being cast on the concept that there was a neutrophil chemotactic defect in periodontitis patients. Once it was realized that neutrophils were primarily protective and that severe periodontal destruction occurred in the absence of these cells, attention swung back to the role of lymphocytes and in particular the regulatory role of T-cells. By this time in the early 1990s, while the roles of interleukin (IL)-1, prostaglandin (PG) E2, and metalloproteinases as the destructive mediators in periodontal disease were largely understood, the control and regulation of these cytokines remained controversial. With the widespread acceptance of the Th1/Th2 paradigm, the regulatory role of T-cells became the main focus of attention. Two apparently conflicting theories have emerged. One is based on direct observations of human lesions, while the other is based on animal model experiments and the inability to demonstrate IL-4 mRNA in gingival extracts. As part of the "Controversy" series, this review is intended to stimulate debate and hence may appear in some places provocative. In this context, this review will present the case that destructive periodontitis is due to the nature of the lymphocytic infiltrate and is not due to periodic acute exacerbations, nor is it due to the so-called virulence factors of putative periodontal pathogens.
(I) Introduction
It is now generally accepted that periodontal disease results from the inflammatory response to bacteria in dental plaque. However, the innate susceptibility of the patient determines the ultimate outcome of the disease process. In other words, it is the nature of the inflammatory response which determines the destructive character of the disease. With increased bacterial challenge, bacterial products interact with the gingival epithelium to induce the expression of adhesion molecules and the production of pro-inflammatory cytokines and chemokines. Blood vessels underlying the affected epithelium show increased permeability and express adhesion molecules, and a gradient of chemoattractant signals guides leukocytes into the gingival tissues. Neutrophils migrate through the junctional epithelium and into the gingival sulcus. However, with the continued presence of bacteria, the formation of an inflammatory infiltrate in the connective tissues consisting predominantly of lymphocytes (T-cells) and macrophages takes place. Subsequently, in a susceptible person, this adaptive response still does not contain the microbial challenge leading to a change in the nature of the inflammatory response (B-cells/plasma cells) which results in either protective antibody and subsequent control of the infection or non-protective antibodies and connective tissue destruction and bone loss. Resident epithelial cells, endothelial cells, and fibroblasts respond to agents such as bacterial lipopolysaccharide (LPS), IL-1, tumor necrosis factor-alpha (TNF-α), and prostaglandins, particularly PGE2, and participate in the tissue destruction. Fibroblasts normally produce several collagens; however, in active periodontitis, the genes for collagens are turned off, as are the genes for tissue inhibitors of metalloproteinases, and the genes for matrix metalloproteinases are turned on, resulting in destruction of the extracellular matrix to make way for the increasing inflammatory cell infiltrate (reviewed in Page et al., 1997).
Page et al. (1997) have hypothesized that disease progression is due to a combination of several factors, including the presence of periodontopathic bacteria, high levels of pro-inflammatory cytokines, matrix metalloproteinases, and PGE2, and low levels of IL-10, TGF-β, and tissue inhibitors of metalloproteinase. In this concept, it is clear that the balance of cytokines determines whether tissue destruction occurs or homeostasis is maintained (Page et al., 1997). In non-susceptible individuals, neutrophils and cell-mediated immunity will limit the extent of attachment loss. However, in susceptible individuals and in the presence of defined periodontopathic bacteria, such as Porphyromonas gingivalis, Actinobacillus actinomycetemcomitans, or Bacteroides forsythus, clearance by neutrophils will be averted and disease progression will occur. The nature of the adaptive immune response is under the control of T-cells which regulate B-cell/plasma cell differentiation and antibody production. Successful neutrophil clearance may depend upon the presence of interferon-gamma (IFN-γ) and may be further enhanced by protective antibodies which in turn are controlled by the types of cytokines produced by T-cells (reviewed in Page et al., 1997). Therefore, it is clear that cytokines are fundamental in determining whether periodontal disease progresses or remains stable, and the cytokines present in periodontal disease tissue are determined by the nature of the lymphocytic response.
(II) Innate Immunity
(A) Neutrophils in periodontal disease
Phagocytic cells such as neutrophils and macrophages constitute the first line of defense against bacterial infection. Neutrophils have been described as playing a major role in periodontal disease (Dennison and Van Dyke, 1997), with both protective and destructive influences being suggested. Much of the evidence suggesting a protective function comes from the observation that individuals who have neutrophil disorders such as cyclic neutropenia (Cohen and Morris, 1961), Chédiak-Higashi syndrome (Hamilton and Giansanti, 1974), and leukocyte adhesion deficiency syndrome (Springer et al., 1984; Waldrop et al., 1987) have an increased susceptibility to periodontal destruction. Further, MacFarlane et al. (1992) reported significantly impaired phagocytosis due to a decreased rate of adhesion and opsonization by neutrophils from patients with refractory periodontitis compared with healthy patients. In the sulcus, neutrophils form a barrier between the epithelium and plaque (Attström and Schroeder, 1979), which may prevent bacterial invasion of the epithelium and underlying connective tissue (Hemmerle and Frank, 1991).
Neutrophils therefore can minimize the destructive effects of plaque bacteria (Dennison and Van Dyke, 1997). However, bacteria such as P. gingivalis are able to evade host innate immune responses (reviewed in Darveau et al., 1997). P. gingivalis, for example, does not stimulate the expression of the neutrophil binding adhesion molecule E-selectin on endothelial cells, thus inhibiting their migration from the circulation into the tissues. It also blocks the expression of this adhesion molecule by other Gram-negative bacteria and their LPS (Darveau et al., 1995). Inhibition of neutrophil migration into P. gingivalis-induced lesions has been demonstrated in our mouse model (Bird et al., 1995). Further, P. gingivalis-induced blocking of neutrophil transmigration through oral epithelium has been reported to be due to inhibition of epithelial cell production of the chemokine IL-8 (Madianos et al., 1997).
Bacteria including P. gingivalis produce proteases which can cleave complement and immunoglobulins, thus preventing opsonization and subsequent neutrophil killing of invading bacteria. Gingipain-R, the major arginine-specific proteinase from Porphyromonas gingivalis, has been shown to cleave a model peptide G-protein-coupled receptor found on the surface of neutrophils (Lourbakos et al., 1998). Certain strains of P. gingivalis produce enzymes which can cleave the Fc regions of bacterial-bound IgG, preventing phagocytosis, and can also cleave the C3b complement component, thereby causing evasion of neutrophil clearance (Sundqvist et al., 1984; Schenkein, 1988; Schenkein et al., 1995). Additionally, since protective antibody production enhances neutrophil clearance (Page et al., 1997), it is possible that P. gingivalis may induce antibodies with poor antibacterial properties (Slots, 1999). If the antibodies are not protective and neutrophils are inhibited, bacteria will escape clearance. A. actinomycetemcomitans also produces a protein which inhibits neutrophil chemotaxis and H2O2 production (Ashkenazi et al., 1992a,b). In addition, it also produces a cytolytic leukotoxin which lyses susceptible target cells, including neutrophils, monocytes, and T-cells (Taichman et al., 1980, 1991; Mangan et al., 1991). Despite this, however, the bacteria per se do not cause tissue destruction in periodontal disease. Rather, it is the nature of the inflammatory response to these bacteria that results in tissue destruction.
A regulatory role for neutrophils in periodontitis has been postulated (Seymour et al., 1993) based on the observation that they secrete a range of cytokines including IL-1 and the IL-1 receptor antagonist (Lloyd and Oppenheim, 1992). This is supported by a study which showed that peripheral blood neutrophils stimulated with a range of periodontopathic bacteria did not produce IL-1 but rather an IL-1 inhibitor, although the nature of this inhibitor was not investigated at the time (Yamazaki et al., 1989). The ability of the innate immune system to regulate the adaptive immune response via the production of cytokines such as the IL-1 receptor antagonist and IL-12 is being recognized. A strong innate immune response may, on the one hand, clear the periodontopathic bacteria and, on the other, determine the nature of the lymphocytic response, which may subsequently result in a stable or progressive form of the disease.
(B) Macrophages in periodontal disease
Macrophages are important mediators of inflammation in the connective tissue infiltrate, where they produce several cytokines and also present antigens to T-cells (Dennison and Van Dyke, 1997). Activated macrophages are essential for innate resistance to intracellular infection. They produce pro-inflammatory cytokines which enhance phagocytosis and in most cases result in the successful elimination of the pathogen (Trinchieri, 1997). Complete elimination of pathogens, however, involves antigen-specific T- and B-cells. The mechanisms of innate and adaptive immune responses are hence seen to be inter-dependent (Trinchieri, 1997). Cells of the innate response influence T-cell differentiation by influencing the cytokine milieu in the tissues or draining lymph nodes where antigen-specific T-cells expand in response to antigen presentation. A high level of IL-12 during T-cell expansion will influence Th1 differentiation, while IL-4 is necessary for Th2 cells (Mosmann and Coffman, 1989; Scott, 1993).
Few studies have been reported on macrophages in periodontal disease. However, while A. actinomycetemcomitans and P. gingivalis have been shown to activate monocytes and macrophages and stimulate the secretion of pro-inflammatory and tissue-destructive mediators (Zadeh et al., 1999), a recent report has shown that there is no increase in macrophage numbers and little evidence of macrophage activation in advanced periodontitis compared with minimally inflamed tissues (Chapple et al., 1998). This is supported by reports demonstrating negative effects by periodontal pathogens on monocytes/ macrophages. For example, P. gingivalis has been shown to affect macrophage migration and activation by inhibiting the production of one of the major monocyte/macrophage chemokines, monocyte chemoattractant protein-1 (MCP-1) (Gemmell et al., 2000a). This suggests that macrophages may have protective effects in the stable lesion which are abrogated in the advanced destructive lesion. P. gingivalis, for example, has also been demonstrated to induce the production of IL-1-β in B-cells, rather than in monocytes, from periodontitis patients (Gemmell and Seymour, 1998). This suggests that the major source of IL-1 in periodontal disease may be lymphocytic rather than macrophages, further supporting the concept that destructive periodontitis is determined by the nature of the lymphocytic response.
(III) Susceptibility to Periodontal Disease
(A) Environmental factors
While periodontopathic bacteria and the inflammation they provoke are essential for disease progression, environmental risk factors such as tobacco smoking, psychosocial stress, and systemic diseases such as diabetes modify the host response and may be major determinants of the enormous variation in susceptibility (Page et al., 1997). These factors may modify the pathways by which bacteria cause inflammation and hence modify disease progression, severity, and outcome (Page et al., 1997). It is generally agreed that smoking is a risk factor for periodontal disease (Grossi et al., 1994, 1995, 1996), exerting both local and systemic effects. However, in a recent study, smoking has been shown to have little effect on disease progression but rather to have a highly significant effect on disease regression or healing, such that the increased disease expression observed in smokers is likely to be due to the inability to heal and the cumulative effect of disease over time (Faddy et al., 2000).
(B) Genetic factors
Innate susceptibility to periodontal disease is influenced by host genotype (Michalowicz, 1994). The relative contributions of genetic factors to clinical measures of periodontal disease have been studied in reared-together twins and monozygous twins reared apart (Michalowicz et al., 1991). This study suggested that from 38 to 82% of the population variance for the periodontal measures of disease could be attributed to genetic factors.
Genetic polymorphisms in Fc receptors on phagocytic cells have been suggested to be significant in determining susceptibility to bacterial infections (Sanders et al., 1995). The Fc receptor for IgG2 is FcγRIIa, and individuals with low-affinity receptors have reduced IgG2-mediated phagocytosis of encapsulated bacteria and are susceptible to a variety of bacteria, including meningococcal infections (Bredius et al., 1994; Sanders et al., 1994). However, whether this is also true for individuals with chronic periodontitis has not yet been demonstrated (Page et al., 1997).
Several studies have reported a predominance of serum IgG2 antibodies in response to P. gingivalis (Lopatin and Blackburn, 1992; Whitney et al., 1992; Polak et al., 1995; Wilton et al., 1993). IgG2 antibodies are the primary immunoglobulin subclass produced in response to bacterial carbohydrates and LPS and lack strong complement fixation and opsonic properties. In periodontal disease subjects, a humoral response involving IgG2 may be ineffective in clearing P. gingivalis, depending upon the nature of the Fc receptor. One study of P. gingivalis antibodies in groups of subjects with and without periodontitis and with and without P. gingivalis infection showed that IgG2 anti-P. gingivalis antibodies were significantly higher in the group of individuals who did not have P. gingivalis in their plaque samples and did not have periodontitis, compared with individuals with P. gingivalis in their plaque (Pietrzak et al., 1998). This suggests that IgG2 antibodies may have resulted in clearance of the organism, although the Fc receptor polymorphism status of these patients was unknown.
Cytokine polymorphisms are currently receiving a great deal of attention, and there is increasing evidence that genetic variation within the TNF locus is an important factor in determining susceptibility to several diseases, including malaria, leishmaniasis, and coeliac disease (McGuire et al., 1994; Wilson et al., 1995, 1997; Manus et al., 1996). Recently, TNF genotypes of 3 bi-allelic polymorphisms were determined in adult periodontitis patients and compared with those in healthy control subjects (Galbraith et al., 1998). The level of TNF-α production by oral neutrophils correlated with the TNF-α 308 genotype in the periodontitis patients, with increased production found in patients with the T1,2 genotype. This association was found only in patients with advanced disease, suggesting that the TNF-α genotype may be a risk factor for disease severity in periodontal disease.
Similarly, IL-1 polymorphisms have been claimed to be a risk factor for severe periodontal disease, with genotype-positive individuals having a 20 times increased risk of developing severe periodontitis after the age of 40 than those individuals who are genotype-negative (Kornman et al., 1997). The polymorphism of the composite periodontitis-associated genotype linked with expression of high levels of IL-1 has been examined in a group of periodontitis patients, with 7/22 individuals being positive for this genotype (Engebretson et al., 1999). The positive and negative groups were comparable in terms of existing periodontitis and age, and while IL-1-β levels in the gingival crevicular fluid were higher in the positive group, a reduction in IL-1-β levels after periodontal treatment occurred only in the negative group. There was also a trend toward higher IL-1-β levels in gingival biopsies of the positive group. Recently, the frequency of IL-1-β genotypes including allele 2 of the IL-1-β+3953 restriction fragment length bi-allelic polymorphism was shown to be significantly increased in patients with adult periodontitis compared with individuals with early and moderate disease (Gore et al., 1998). Further, allele 2 was associated with increased production of IL-1-β by activated peripheral blood neutrophils isolated from patients with advanced disease. In a more recent study, it was shown that the IL-1 allele 2 composite phenotype was a contributing but non-essential risk factor for periodontal disease progression over a five-year period (Cullinan et al., 2001).
(IV) Adaptive Immunity
In 1965, Brandtzaeg and Kraus demonstrated the presence of immunoglobulin-producing plasma cells in the gingival tissues of patients with periodontal disease. This was the first direct evidence that adaptive immune mechanisms may play a role in the pathogenesis of periodontal inflammation.
Ivanyi and Lehner (1970) used peripheral blood lymphocyte transformation assays to highlight a role for cell-mediated immunity in periodontal disease. Subsequently, a great many studies using a variety of lymphokine assays confirmed that cell-mediated responses did play some part in pathogenesis, but the exact role was unclear.
Direct immunohistologic investigation of the tissues throughout the 1970s and 1980s (Mackler et al., 1977; Seymour and Greenspan, 1979; Lindhe et al., 1980; Seymour et al., 1983, 1988; Reinhardt et al., 1988) seemed to confirm the hypothesis that a change from gingivitis to periodontitis involved a shift from a predominantly T-cell lesion to a B-cell/plasma cell lesion (Seymour et al., 1979). This hypothesis was also supported by functional studies, albeit using peripheral blood cells, which showed that suppressed cell-mediated immunity in advanced periodontitis subjects (Ivanyi and Lehner, 1970; Boyatzis and Seymour, 1986) could be reversed following treatment (Evans et al., 1989). Other studies also demonstrated lymphocyte suppression by periodontopathic bacteria. Low responses to P. gingivalis, Treponema denticola, and Capnocytophaga ochracea were found in both healthy and periodontitis peripheral blood lymphocytes compared with other oral bacteria (Stashenko et al., 1983), and in another study, P. gingivalis reduced the response of non-mitogen-stimulated cells (Shenker and Slots, 1989; Gemmell and Seymour, 1992). A. actinomycetemcomitans was also shown to suppress the peripheral blood lymphocyte response to mitogens (Shenker et al., 1982a), and depletion of CD8 cells was found to abrogate this response, suggesting the induction of a CD8 suppressor factor (Shenker et al., 1982b).
In terms of the transition from gingivitis to periodontitis, several studies have reported a decreased CD4/CD8 ratio (Taubman et al., 1984a; Cole et al., 1987; Stoufi et al., 1987; Okada et al., 1988; Wang et al., 1989) in periodontal lesions, while functional studies have reported a reduced autologous mixed-lymphocyte reaction (Cole et al., 1987). Taken together, these studies have suggested a local immunoregulatory imbalance. It is now generally agreed that both T- and B-cells are present in periodontal disease tissues (Zadeh et al., 1999). It is also clear that the majority of T-cells are activated memory/primed cells (Okada et al., 1988; Reinhardt et al., 1988; Takeuchi et al., 1991; Gemmell et al., 1992; Yamazaki et al., 1993). Indeed, both T- and B-cells extracted from gingival tissues have been reported to be at a more advanced stage of the cell cycle than peripheral blood T- and B-cells, indicative of activation within the tissues (Gemmell and Seymour, 1992).
(A) B-cells in periodontal disease
Polyclonal B-cell activation has been cited as being significant in the pathogenesis of periodontal disease. Sonic extracts of periodontopathic bacteria were first shown to induce polyclonal B-cell activation of peripheral blood lymphocytes, as assessed by the plaque-forming cell response to sheep red blood cells (Bick et al., 1981; Donaldson et al., 1982) and by the production of antibodies to several different organisms as well as specific antibody to the stimulating organism (Mangan et al., 1983; Carpenter et al., 1984). Several studies have shown that this polyclonal B-cell response to oral organisms is dependent on the presence of T-cells (Donaldson et al., 1984; Okada et al., 1987; Ito et al., 1988). While most of these studies examined peripheral blood responses, an in situ study recently reported that local proliferation of B-cells does not occur in periodontitis tissues, although plasma cells showed strong synthetic activity (Takahashi et al., 1996). However, Gemmell and Seymour (1991) previously demonstrated that B-cells extracted from periodontitis tissues do have a more activated phenotype than cells from gingivitis tissues or peripheral blood. Murine models have been used to demonstrate the induction of polyclonal B-cell activation by A. actinomycetemcomitans and P. gingivalis (Inada et al., 1994; Watanabe et al., 1996). However, when T- and B-cells from mice immunized with a 40-kDa immunodominant outer membrane antigen of P. gingivalis were examined in vitro, the secondary antibody response required the presence of B-cells, CD4+ T-cells, and H-2 restricted, Ia+ antigen-presenting cells (APC) (Oshikiri et al., 1994). The antibody response to the 40-kDa P. gingivalis antigen, therefore, was generated in an antigen-specific manner and was not the result of polyclonal B-cell activation.
The inability of specific antibodies to eliminate the causative organisms of periodontal disease could be due to several factors, including poor antigenicity of the virulence determinants and elicitation of antibodies with poor antibacterial properties (Slots, 1999). High titers of specific antibody levels to P. gingivalis (Ling et al., 1993; Wilton et al., 1993; Meghji et al., 1995) and A. actinomycetemcomitans (Saito et al., 1993; Meghji et al., 1995; Engström et al., 1999) have been reported in serum and gingival crevicular fluid, although many studies report conflicting results. Positive correlations between elevated IgG levels to P. gingivalis and A. actinomycetemcomitans have been shown in gingivitis associated with both puberty and adult periodontitis (Nakagawa et al., 1994), while a negative relationship between anti-A. actinomycetemcomitans antibodies in the serum of periodontitis patients and the burden of A. actinomycetemcomitans in infected sites has also been reported (Ebersole et al., 1994). Another study demonstrated no difference in the levels of anti-P. gingivalis antibodies in the gingival crevicular fluid of periodontitis patients and healthy control subjects, although there were moderate to strong correlations with serum antibody levels (Baranowska et al., 1989).
Serum antibody changes have been suggested to reflect the nature of the organism load (Taubman et al., 1992), and elevated levels of anti-P. gingivalis IgG antibodies have been linked to the presence of marked periodontal lesions (Gmür et al., 1986), while levels of antibodies in the gingival crevicular fluid have been cited to be an inverse of the number of organisms at the particular site of sampling (Kinane et al., 1993; Ebersole et al., 1995). Kinane et al. (1993) demonstrated a correlation of specific serum antibody titers to P. gingivalis and A. actinomycetemcomitans with mean gingival crevicular fluid titers, suggesting protective humoral immune responses, although the IgG titers to P. gingivalis in sites with higher probing depths were lower, so that patients with greater inflammation had lower IgG levels. In another group of patients, Mooney and Kinane (1997) found that anti-P. gingivalis IgG antibody levels in periodontitis sites were lower than in gingivitis sites in the same subjects, suggesting that a failure in local antibody production may contribute to the change from gingivitis to periodontitis lesions. Reinhardt et al. (1989) measured IgG subclasses in the gingival crevicular fluid of periodontally active and stable or healthy sites. The mean IgG1 and IgG4 concentrations were higher in the fluid from active periodontitis sites than in stable sites, suggesting that these antibodies were ineffective in eradicating the bacteria. At the same time, the mean levels were generally greater than in serum, especially for IgG4, further suggesting that this subclass may be a useful indicator for immunological changes which occur in active periodontal disease.
Humoral immune response studies have also focused on the determination of immunodominant antigens and a study of their functions. However, different patterns of immunoreactivity with periodontopathic bacteria have been demonstrated for A. actinomycetemcomitans (Califano et al., 1991, 1992; Engström et al., 1993; Ebersole et al., 1995) and for P. gingivalis (Curtis et al., 1991; Kurihara et al., 1991; Polak et al., 1995). Since these responses are regulated by immunoregulatory genes, it may be that antibody responses are protective in one individual but not in another (Offenbacher, 1996).
IgG subclasses and avidity have also been implicated in periodontal disease progression; however, the functional characteristics which may help to define susceptibility to the disease are still to be ascertained (Ishikawa et al., 1997). Underwood et al. (1993) suggested that high levels of anti-A. actinomycetemcomitans antibodies with high avidity may confer greater resistance to continued or repeated infection. Mooney et al. (1993) showed that patients who did not experience attachment loss during a three-month monitoring period had anti-P. gingivalis antibodies with higher avidity than those patients who did experience attachment loss. Further, Lopatin and Blackburn (1992) showed that anti-P. gingivalis and anti-P. gingivalis-LPS antibodies in periodontitis patients had significantly lower avidity than antibodies to streptokinase and tetanus toxoid, and while antibodies of the IgG1 subclass were of relatively high avidity, those in the IgG2 subclass, which were the predominant IgG subclass, were of significantly lower avidity. The relationship between titers and avidities of serum antibodies to P. gingivalis and A. actinomycetemcomitans and to clinical parameters of periodontal disease severity and the level of infection with the homologous organism has been examined in a group of periodontitis and gingivitis patients (Lamster et al., 1998). There was a negative correlation between the mean probing depths and antibody titer and avidity to A. actinomycetemcomitans and to infection with A. actinomycetemcomitans, while there was a positive correlation with respect to P. gingivalis. It was concluded that the development of an antibody response to A. actinomycetemcomitans appears to protect individuals from infection, whereas the antibody response to P. gingivalis was not able to eliminate the infection. Non-protective low-avidity anti-P. gingivalis antibodies may be incapable of effectively mediating a variety of immune responses (Lopatin and Blackburn, 1992; Whitney et al., 1992). The general inability to demonstrate immune complexes in the gingival sulcus (Clagget and Page, 1978) supports the notion that there is a compromised ability to eliminate or reduce the numbers of micro-organisms and their products from the gingival sulcus (Lopatin and Blackburn, 1992). The production of anti-P. gingivalis antibodies with different avidities in various forms of periodontal disease has been suggested to reflect the quality of the humoral response, which may affect progression of the disease (Mooney and Kinane, 1994). Wilton et al. (1993) reported that low titers to organisms such as P. gingivalis in healthy individuals were of low avidity.
The immune responses to self-antigens such as collagen type I, a major component of the periodontium, have also been suggested as a pathogenic pathway. High titers of anti-collagen type I antibody are found in the sera (Hirsch et al., 1988) and anti-collagen type I specific T-cell clones can be identified in the inflamed gingival tissues of periodontitis patients (Wassenaar et al., 1995). Sugawara et al. (1992) demonstrated that the auto-reactive CD5+ B-cells increased in periodontitis lesions and the CD5+ B-cells produced more IgM and IgG antibodies to collagen in vitro than did CD5-B-cells (Sugawara et al., 1992). Although the mechanisms that induce an immune response to self-components are not fully elucidated, molecular mimicry has been cited to explain the linkage between infection with bacteria and subsequent autoimmune mechanisms (Oldstone, 1987). Antibody responses to heat-shock protein 60 (hsp60) in particular are thought to be important. It has been reported that GroEL-like protein belonging to the hsp60 family is expressed by periodontopathic bacteria such as P. gingivalis (Hotokezaka et al., 1994; Maeda et al., 1994) and A. actinomycetemcomitans (Nakano et al., 1995), while Tabeta et al. (2000) have demonstrated that serum antibodies to P. gingivalis GroEL and human hsp60 are elevated in periodontitis patients compared with healthy control subjects (Tabeta et al., 2000). They also demonstrated that these antibodies cross-reacted with one another and concluded that the immune response to bacterial hsp60 may be an important pathogenic mechanism in periodontitis.
During the chronic phase of the disease, the antibody response has been suggested to be generally protective, to facilitate bacterial clearance, and to arrest disease progression (Offenbacher, 1996). Serum from patients with severe periodontitis containing high titers of anti-P. gingivalis antibodies completely inhibited in vitro bone resorption, whereas serum from patients with low titers failed to inhibit this bone resorption, confirming a possible protective role for specific antibodies in periodontitis (Meghji et al., 1993). Other studies suggesting a role for protective antibodies include the demonstration of anti-P. gingivalis protease antibodies which occur late in periodontitis infections and which can block the anti-opsonizing activity against C3 and IgG (Cutler et al., 1993). An increased capacity of serum to opsonize P. gingivalis has been shown to be a distinctive feature in patients with past destructive periodontal disease (Wilton et al., 1993). Opsonic IgG antibodies to A. actinomycetemcomitans which may facilitate neutrophil-mediated phagocytosis and be protective against this periodontopathic organism have also been demonstrated (Baker and Wilson, 1989; Underwood et al., 1993). Repeated infection with A. actinomycetemcomitans has been shown to elicit an anti-leukotoxin antibody which protects neutrophils from the leukocidal activity of the leukotoxin (Underwood et al., 1993). Tsai et al. (1981) showed that the majority of sera from juvenile periodontitis patients contain antibodies which neutralize A. actinomycetemcomitans leukotoxin, while sera from individuals without disease or those with other types of periodontal disease usually amplified rather than inhibited the reaction to the A. actinomycetemcomitans leukotoxin. On the other hand, however, Cutler et al. (1991) reported that only 3/17 serum samples from several adult periodontitis patients with elevated IgG to P. gingivalis A7436 were opsonic for this particular strain. In another study, antibody levels to subgingival plaque micro-organisms were found to be a predictor of bone loss in an elderly group of patients with anti-P. gingivalis antibodies to two strains of P. gingivalis having significant correlations with loss of alveolar bone (Wheeler et al., 1994).
(B) T-cells in periodontal disease
It would appear, therefore, that locally produced specific antibodies may be able to clear the causative organism in some people, while in others polyclonal B-cell activation and the production of low-avidity specific antibodies may not clear the infection, and disease progresses. Offenbacher (1996) suggests that if the antibody/neutrophil response does not result in clearance, the outcome of the monocyte/lymphocyte challenge is the secretion of catabolic cytokines and inflammatory mediators such as IL-1, IL-6, TNF-α, and PGE2, which induce connective tissue and bone loss. As the microbial challenge increases, and with repeated challenge, the antibody response may become protective and enable neutrophil clearance to occur, and the infection reaches homeostasis. Such a concept, however, is not in accord with the observation that the monocyte/lymphocyte infiltrate occurs early in disease formation (Page and Schroeder, 1976; Seymour et al., 1988) followed by the B-cell/plasma cell lesion which is characteristic of the advanced forms of the disease (Page and Schroeder, 1976). An alternative hypothesis, therefore, would be that failure of the innate immune response to clear the infection, even in the presence of stimulating cytokines such as IFN-γ, leads to the development of a B-cell/plasma cell lesion. If as a result specific antibodies with high avidity are formed, the infection is cleared and the disease does not progress. If on the other hand polyclonal B-cell activation and non-specific and/or low-avidity specific antibodies are produced, the infection is not cleared. Continued B-cell activation leads to the production of high levels of IL-1, and as a result, tissue destruction ensues. This hypothesis is consistent with the normal functioning of the immune system in bacterial infections, with the observation that B-cells are a potent source of IL-1, that macrophages are not a dominant feature of the advanced lesion, and that suppressed cell-mediated immunity is associated with advanced periodontitis.
Clearly, whichever hypothesis is correct, T-cells must play a fundamental role. T-cells are the dominant cell type in the cell-mediated (macrophage/lymphocyte) response and are necessary for both specific antibody production and polyclonal B-cell activation.
Immunohistological studies have clearly established that a T-cell/macrophage lesion identical to a delayed hypersensitivity reaction (Poulter et al., 1982) occurs within 4 to 8 days of plaque accumulation in an experimental gingivitis study (Seymour et al., 1988). This is synonymous with the early lesion of Page and Schroeder (1976) and with the putative stable lesion (Seymour et al., 1979). The expression of HLA-DR and DQ by these cells indicates that they are activated, but the lack of CD25 expression would indicate that they are not proliferating locally within the tissues (Seymour et al., 1988). The striking similarities between this early/stable periodontal lesion and delayed-type hypersensitivity prompted the suggestion that cells with a Th1 cytokine profile are the major mediator. Such a concept is consistent with the proposal that a strong innate immune response leads to the production of IL-12, which in turn leads to this Th1 response. The production of IFN-γ then enhances the phagocytic activity of both neutrophils and macrophages and hence containment of the infection. However, the stable lesion persists due to the continual formation of the plaque biofilm (Gemmell and Seymour, 1994).
The dominance of B-cells/plasma cells in the advanced/ progressive lesion would suggest a role for Th2 cells. Clearly, if the innate response is poor, low levels of IL-12 would be produced and a poor Th1 response may occur which may not then contain the infection. Mast cell stimulation and the subsequent production of IL-4 would encourage a Th2 response, B-cell activation, and antibody production. If as noted above these antibodies are protective and clear the infection, the disease will not progress, but if on the other hand they are non-protective, the lesion will persist, and continued B-cell activation would result in large amounts of IL-1 and hence tissue destruction (Seymour et al., 1993; Gemmell and Seymour, 1994, 1998).
To test this hypothesis, several investigators have attempted to delineate the Th1/Th2 profile in periodontal disease. However, results are difficult to interpret due to differences in material examined and methodologies used. Cytokines have been studied in cells in situ, cells extracted from gingival tissues, peripheral blood mononuclear cells, T-cell lines, and clones as well as purified cell populations. A variety of techniques has also been used, including flow cytometry, enzyme-linked immunosorbent assay (ELISA), in situ hybridization, and reverse-transcriptase/polymerase chain-reaction (RT-PCR). Additionally, bacterial components including sonicates, heat- and formalin-killed cells, outer membrane components, and purified antigens have all been used to stimulate cells in vitro, making comparisons of cytokine profiles difficult (Zadeh et al., 1999). It is likely, as indicated above, that different T-cell subsets predominate at different phases of disease, and the inability to determine disease activity clinically is a major limitation in all these studies.
Several investigators have reported decreased Th1 responses in periodontal disease (Table). Pilon et al. (1991) demonstrated lower levels of IL-2 in the gingival crevicular fluid of periodontitis sites compared with healthy sites, and Fujihashi et al. (1991) have shown that gingival mononuclear cells from adult periodontitis patients produce IL-4 and IL-5 but not IL-2. Peripheral blood mononuclear cells from periodontitis patients stimulated with mitogens resulted in reduced IFN-γ secretion and mRNA expression of IFN-γ and IL-2. At the same time, significantly higher levels of IL-5 and GM-CSF were observed (Sigusch et al., 1998). Significantly less IL-2 activity was also found in peripheral blood mononuclear cell cultures stimulated with P. gingivalis and another oral Gram-negative anaerobe, Fusobacterium nucleatum, compared with unstimulated cultures (Gemmell and Seymour, 1994). In this study, IFN-γ as measured by an ELISA could not be detected in cultures containing both bacteria; furthermore, IFN-γ was demonstrated in only 10/27 gingival mononuclear cell culture supernatants. A recent in vivo study has confirmed these observations, showing that priming of mice with F. nucleatum prior to challenge with P. gingivalis resulted in a polarized Th2 response, while P. gingivalis alone resulted in a Th1 response (Choi et al., 2000). This important paper highlights the complexities involved in periodontal disease and the dangers of using single organisms in highly artificial settings.
Increased Th2 responses in periodontitis have also been reported (Table). Memory T-cells from the peripheral blood of adult periodontitis patients with high anti-P. gingivalis titers stimulated in vitro with P. gingivalis have been shown to produce higher amounts of IL-4 than cells from healthy subjects (Aoyagi et al., 1995). In this study, no IL-4-producing memory T-cells were detected in healthy gingival tissues, and a larger proportion of peripheral blood memory T-cells from patients in whom high frequencies of IL-4 producing cells were identified in the lesion produced IL-4 following stimulation with antigen. Yamazaki et al. (1994a) demonstrated an increased percentage of IL-4+ cells proportional to an increasing B-cell/T-cell ratio. IL-4 was the prominent cytokine in periodontally affected tissues compared with IL-2, IFN- , and IL-6. Another study suggested a role for IL-4 and Th2 responses in periodontitis lesions by the demonstration of concentrations of IgG4 many times higher in sites of active periodontitis than in serum, as well as significantly elevated concentrations compared with stable lesions (Reinhardt et al., 1989). A bias toward Th2-type cytokines in periodontal disease progression was also indicated when cytokine analysis of cells in inflamed gingiva by in situ hybridization showed that the density of cells expressing IL-1- , IL-4, and IL-5 mRNA was higher in periodontitis than in gingivitis (Tokoro et al., 1997). Further, cell dot-blot analysis of cytokine-producing gingival mononuclear cells showed that a higher percent of non-stimulated periodontal disease cells were IL-4+ (Manhart et al., 1994). Analysis of IL-2/IL-4 ratios revealed significantly lower ratios for cells derived from periodontitis tissues compared with cells from gingivitis tissues. An immunohistochemical study demonstrated a significantly higher level of IL-4-producing cells in periodontal lesions in comparison with gingivitis tissues, and although this was also the case for IL-6-producing cells, the results were not significant (Yamazaki et al., 1994b). In this study, Th2-type cells were shown to accumulate in periodontitis. In a recent study, peripheral blood mononuclear cells isolated from P. gingivalis-positive gingivitis and periodontitis subjects were stimulated with P. gingivalis outer membrane antigens. IL-4+ T-cells predominated over IFN-γ+ and IL-10+ cells, although there were no differences in the percentages of positive cells in comparison with cells stimulated in the absence of P. gingivalis antigens (Gemmell and Seymour, 1998), suggesting a non-specific effect perhaps due to the very low numbers of P. gingivalis-specific precursor T-cells present in the peripheral circulation of both healthy and diseased individuals (Mahanonda et al., 1991). Taken together, however, these data seem to support the hypothesis that Th1 cells are associated with the stable lesion and a Th2 response with disease progression.
In contrast to these studies, Ebersole and Taubman (1994) found that IFN-γ message was prominently expressed by diseased gingival tissue cells (Table). Cytokine profiles of cells extracted from six patients were consistent with Th1 cells in that they were IL-2- and IFN-γ-positive but negative for IL-4 and IL-5. A further sample had message for IL-2 and IL-5, consistent with Th0 cells. The investigators' inability to detect IL-4 mRNA in these experiments could have been due to its very short half-life rather than to its absence from the tissues. As with other studies, disease activity was not clearly defined, such that, again, interpretation is difficult. Another study comparing the local and systemic responses in periodontitis patients with so-called terminal dentition periodontitis demonstrated reduced Th2 responses. Levels of PGE2, IL-1-β, and IL-2 in the gingival crevicular fluid samples were highest, followed by lower levels of TNF-α and IFN-γ and even lower levels of IL-4 and IL-6 (Salvi et al., 1998) (Table). LPS-stimulated monocytes from these patients resulted in significantly elevated levels of PGE2, IL-1-β, and TNF-α compared with cultures of cells derived from control subjects with moderate to advanced disease. Cytokine mRNA expression of Th1 and Th2 cytokines was present in isolated gingival mononuclear cells with low levels of IL-4 and IL-12. While it was concluded that Th1 cytokine levels dominated over the Th2 response in the gingival crevicular samples, and that monocyte activation provided the major source of pro-inflammatory mediators, because different concentrations of different cytokines result in different levels of biological activity, the comparison of different levels of cytokines in the same patient is inappropriate. It would have been of greater value had the levels of the same cytokines been compared in patients from different disease groups. Equally, the ability of peripheral blood monocytes to produce cytokines upon stimulation does not imply that these are the major source of these cytokines in a highly localized lesion as is seen in periodontal disease. Interpretation of this study is therefore difficult.
Other studies have suggested the involvement of Th0 cells in periodontal disease (Table). Fujihashi et al. (1994) extracted mRNA for IFN-γ, IL-6, and IL-13, but not for IL-2, IL-4, or IL-5 in CD4+ T-cells from periodontal disease lesions. Takeichi et al. (1994) showed that IFN-γ and IL-1-β mRNA was expressed by some gingival cells on extraction, indicative of Type 1 cells, and upon stimulation, IL-6 transcripts were also expressed but no IL-2 or IL-2 receptor mRNA could be detected. Yet another study found no skewing of cytokines toward a Th1 or Th2 profile in diseased or healthy tissues, although there was a significantly higher expression of IL-6 and IFN-α mRNA in diseased tissues (Prabhu et al., 1996). Fujihashi et al. (1996) isolated CD4 cells from inflamed gingival tissues and demonstrated two profiles, both of which were positive for IFN-γ, IL-6, and IL-13 mRNA, while one group and not the other was also positive for IL-10. Nakajima et al. (1999) demonstrated that stimulation of peripheral blood mononuclear cells from individuals with periodontitis and gingivitis with P. gingivalis resulted in up-regulation of messenger RNA for IFN-γ and IL-13, while IL-4 and IL-10 were down-regulated, regardless of disease status or the presence of P. gingivalis in plaque samples. Message for several cytokines has been compared in the gingival tissues and peripheral blood of periodontitis patients (Yamazaki et al., 1997). The mean expression of IFN-γ mRNA was higher in the peripheral circulation than in the gingival tissues, while that of IL-10 mRNA was higher in the gingival tissues. However, only 7/16 samples demonstrated a high expression of IL-10, the other samples showing equivalent levels in blood and tissues. IL-12 mRNA was similarly expressed to a higher extent in 6/16 samples, the other samples showing no differences between gingiva and peripheral blood. IL-4 mRNA was weak but detectable in only 3 samples.
Studies on T-cell lines and clones have also demonstrated conflicting results (Table). One such study reported that T-cell lines and clones specific for P. gingivalis resembled Th0 cells, although one CD4+ clone did produce IL-4 and IL-5 mRNA, suggesting a Th2 profile (Karatzas et al., 1996). Flow cytometry and RT-PCR have been used to show that CD4 and CD8 cells in P. gingivalis-specific lines and clones derived from a P. gingivalis-positive gingivitis subject and a P. gingivalis-positive periodontitis patient produced IL-4, IL-10, and IFN-γ (Gemmell et al., 1995). However, in a further study, several lines established from P. gingivalis-positive gingivitis and periodontitis subjects demonstrated highly variable profiles, although the mean results showed that a high percentage of both CD4 and CD8 cells was positive for IFN-γ, with lower percentages of IL-4+ and IFN-γ+ cells. Lines established from the periodontitis subjects demonstrated more variation than lines from the gingivitis individuals, such that IL-4+ and/or IL-10+ T-cells predominated over IFN-γ+ cells in a greater number of lines (Gemmell et al., 1999). Wassenaar et al. (1995) established T-cell clones from the gingival tissues of four patients with chronic periodontitis, although these were raised non-specifically with the use of mitogen and IL-2. Eighty percent of the CD4 clones had Th2 phenotypes producing high levels of IL-4 and low levels of IFN-γ, while the majority of CD8 clones demonstrated a Th0-like pattern producing equal amounts of IL-4 and IFN-γ. A further study demonstrated two subsets of CD8 clones expressing the αβ T-cell receptor, one of which produced high levels of IFN-γ but no IL-4 or IL-5 (Th1) and mediated cytolytic activity, while the other subset produced high levels of IL-4 together with IL-5 and displayed no cytotoxicity. The latter Th2 cells suppressed the proliferative response of cytotoxic CD8 T-cell clones, which could be abolished by the addition of anti-IL-4 antibodies, although IL-4 alone could not induce this suppression. Therefore, CD8 T-cells may participate in the local response by suppressing IFN-γ-producing cells and favoring humoral immune responses (Wassenaar et al., 1996). While it is difficult to interpret all these studies, the pattern seems to be emerging that within the tissue, gingivitis seems to be associated with a Th1 response, while periodontitis is associated with a Th2 response. This is consistent with the histology of these two diseases. In peripheral blood, however, no clear pattern is emerging. The reasons for this remain obscure.
Emanating from studies of cytokines in periodontal disease is the concept that IL-10 may be of fundamental importance in the control of periodontal disease progression. A significant decrease in the percent of IL-10+ CD8 cells in gingival cells extracted from periodontitis tissues in comparison with gingivitis samples has been demonstrated (Gemmell and Seymour, 1998), although Stein and Hendrix (1996) found that gingival mononuclear cells extracted from adult periodontitis patients produced more IL-10 than cells derived from non-inflamed tissue. As well, anti-IL-10 antibodies induced an 80% decrease in the frequency of anti-collagen-secreting cells, and it was suggested that IL-10 in inflamed gingival tissues potentiates a local autoimmune response characterized by an increase in anti-collagen-secreting cell frequency. A role for IL-10 has been suggested by another study which analyzed cytokine expression in CD4+ gingival lymphocytes isolated from inflamed periodontal tissues. As reported by Fujihashi et al. (1996), two distinct profiles were noted. One pattern showed the presence of IFN-γ, IL-6, IL-10, and IL-13 mRNA, while the other pattern was similar with the exception of a lack of IL-10 mRNA. In most cases, IL-2, IL-4, and IL-5 mRNA were not detected (Yamamoto et al., 1997). IL-10 has been demonstrated to inhibit LPS-induced B-cell proliferation in the mouse (Marcelletti, 1996), and decreased IL-10 in periodontitis may possibly allow for continued polyclonal B-cell activation.
Studies on cytokines have led to the formulation of several hypotheses as to which T-cell subsets are associated with periodontitis. Seymour et al. (1993) proposed that due to the shift in lymphocyte populations in the inflammatory infiltrate from predominantly T-cells in gingivitis to an increased proportion of B-cells in periodontitis, susceptibility to periodontal disease progression may involve a predominantly Th2-like response in which T-cells produce the cytokines required for B-cell proliferation and differentiation, leading to polyclonal B-cell activation, the production of elevated levels of non-protective antibodies, and the continued production of B-cell IL-1. Non-susceptibility to periodontal breakdown may involve a predominantly Th1-like response, resulting in T-cell activation, cell-mediated immunity, IFN-γ enhancement of innate immunity, and, if necessary, the production of protective antibodies (Fig., A). On the other hand, Ebersole and Taubman (1994) have formulated their hypothesis (Fig., B) based on their adoptive transfer experiments using the Th2 clone A3. They speculate that Th2 cells abrogate periodontal disease symptoms and Th1 cells enhance disease. Th2 cells could be protective, providing help for specific antibody production which they propose is a key feature of protection against periodontal destruction, while Th1 and CD8+ cells may be destructive via the production of IFN-γ and potential stimulation of macrophage IL-1, leading to bone destruction. Th1 cells therefore exert pro-inflammatory effects leading to tissue injury and immunopathology, while Th2 cells produce cytokines leading to anti-inflammatory functions (IL-4 and IL-10 down-regulate IL-1 production [Essner et al., 1989] and IL-10 increases IL-1 receptor antagonist secretion [Schreiber et al., 1995]) and the down-regulation of Th1 cell-mediated tissue-destroying effects (Del Prete et al., 1993).
Yet another hypothesis has been advanced by Dennison and Van Dyke (1997), who suggest that alterations in the monocyte response may lead to abnormal disease patterns (Fig., C). This is based on studies showing that IL-4 inhibits the monocyte secretion of cytokines and PGE2 (Te Velde et al., 1990; Corcoran et al., 1992). Macrophages play a central role in the immune response to bacteria, by acting as APC to initiate responses, producing pro-inflammatory cytokines and other mediators which induce antibacterial responses by effector cells and by the killing of micro-organisms (Ishikawa et al., 1997). LPS is a component of Gram-negative bacteria, and while several LPS receptors have been identified (Lynn and Golenbock, 1992), binding of CD14 on monocytes, macrophages, and neutrophils mediates cytokine secretion (Wright et al., 1990; Weinstein et al., 1993). CD14, one of the major receptors for P. gingivalis LPS (Shapira et al., 1994), is down-regulated by IL-4 (Lauener et al., 1990), and Ishikawa et al. (1997) have suggested that IL-4 may play a major role in the anti-inflammatory effects of LPS-stimulated monocytes by down-regulating CD14 and cytokine secretion by IL-4. Due to reports demonstrating an absence of IL-4-producing T-cells in periodontitis lesions, Shapira et al. (1992) suggest that a lack of down-regulation of monocytes by IL-4 leads to tissue destruction. In support of this hypothesis, gingival macrophages have been shown to express high levels of IL-4 receptor mRNA compared with peripheral blood monocytes, and when incubated with recombinant IL-4, cell viability was reduced as a result of apoptosis. IL-4 may therefore inhibit the persistence of macrophages in periodontitis, which could lead to decreased tissue destruction (Yamamoto et al., 1996). However, IL-13 has biological activities similar to those of IL-4, and since IL-13 expression has been demonstrated in periodontitis lesions (Fujihashi et al., 1996; Yamazaki et al., 1997), it is possible that it may substitute for IL-4 in periodontitis.
Overall, this hypothesis is not consistent with the observation that relatively few macrophages are present in those advanced lesions showing the most tissue destruction (Chapple et al., 1998). If this hypothesis was correct, one would reasonably expect to find the most numbers of macrophages where the tissue destruction was greatest. The lack of macrophages in these advanced lesions, however, is consistent with the presence of IL-4 (and/or IL-13) and hence of a Th2 response. The source of IL-1 remains to be determined, but again, B-cells would seem to be implicated (Gemmell and Seymour, 1998).
Controversy #5: Th1 cells are associated with the stable lesion, while Th2 cells are associated with the progressive lesion in humans.
(V) Animal Models
Investigators have used animal models to try to obtain a clearer picture of the pathogenesis of periodontal disease, which can often be obscured in studies on humans because of the difficulty in diagnosing susceptible and non-susceptible individuals, of determining disease activity, and because of the different bacterial complexes involved in different people. Animal model studies have provided important information toward building a picture of periodontopathic bacteria-mediated response, particularly the responses to P. gingivalis and A. actinomycetemcomitans, but their extrapolation to human disease always remains speculative.
Several investigators have reported on the effects of immunization of non-human primates with P. gingivalis. Anderson et al. (1995) demonstrated the production of an antibody response which enhanced neutrophil phagocytosis of P. gingivalis. However, an enormous variability in the functional capacity of the serum samples was noted, suggesting that antibody function may be a poor predictor of susceptibility to disease. Another study using Macaca fascicularis demonstrated inhibition of the progression of periodontal tissue destruction (Persson et al., 1994). Immunization with a purified P. gingivalis cysteine protease induced a significantly elevated specific IgG response and fewer alveolar bone density changes compared with sham-immunized control animals (Moritz et al., 1998). However, immunization of these animals with combined P. gingivalis and Prevotella intermedia resulted in significantly greater bone density loss in ligated teeth compared with control animals, so that mixed-bacterial immunization resulted in increased disease (Ebersole et al., 1991). This model has also shown that on the induction of experimental disease, the front of inflammatory cells progresses toward alveolar bone, with associated osteoclast formation (Graves et al., 1998). When IL-1 and TNF-α activity was blocked by local injection of soluble receptors to these cytokines, the progression of the inflammatory cells was inhibited, suggesting that this activity was at least partly dependent on IL-1 and/or TNF-α.
Rodent models have been used extensively to study the immune responses to periodontopathic bacteria or their products, including LPS. Early studies demonstrated that LPS from P. gingivalis strains induced marked mitogenic responses and polyclonal B-cell activation in spleen cells, activated both neutrophils and macrophages, and enhanced IL-1 production by murine peritoneal macrophages (Fujiwara et al., 1988; Isogai et al., 1988). However, Kesavalu et al. (1992) found that immunization with whole cells or LPS of P. gingivalis provided no protection against the lethal effects after challenge with LPS. Additionally, Chen et al. (1990) demonstrated that immunization of mice with P. gingivalis LPS did not result in readily detectable IgG or IgM levels to LPS, nor did it reduce the severity of P. gingivalis infection. Taken together, these results suggest that responses to LPS may not be protective, and that a response to these antigens may not prevent progression of disease.
Immunization with whole cells of P. gingivalis, on the other hand, has been shown to elicit protection in the form of faster healing of lesions and production of specific antibodies (Chen et al., 1990; Genco et al., 1992; Kesavalu et al., 1992). Kesavalu et al. (1992) demonstrated that this protection was primarily due to opsonic antibodies, resulting in phagocytic destruction of virulence components. Using 4 strains of inbred mice, Gemmell et al. (2000b) demonstrated that anti-P. gingivalis antibody levels correlated with lesion recovery after challenge with P. gingivalis.
Synthetic peptides of P. gingivalis fimbriae have been reported to induce both Th1 and Th2 cytokines in vitro with the use of in vivo primed mouse T-cells (Deslauriers et al., 1996). Further, mice immunized with recombinant fimbrillin and 2 synthetic peptides induced specific antibodies to fimbrillin, and immunization with one of the peptides, PgF-P8, protected against a lethal injection of P. gingivalis, although the degree of protection depended on the time of immunization before challenge. However, no correlation could be found between protection and in vivo local cytokine production, specific antibody levels, or the isotype of the anti-PgF-p8 antibodies produced (Deslauriers et al., 1996).
Responses of mice to co-infection have been reported. Mice infected with P. gingivalis together with F. nucleatum had significantly greater skin lesions than those infected with P. gingivalis alone, while active immunization with P. gingivalis protected against challenge with both organisms (Ebersole et al., 1997). Protection in this study correlated with levels of specific serum antibodies. Chen et al. (1996) reported that immunization of A. actinomycetemcomitans together with P. gingivalis resulted in first- and second-degree lesions compared with first-degree lesions only, which followed immunization with A. actinomycetemcomitans alone. The serum anti-P. gingivalis response was higher in mice injected with both organisms than in P. gingivalis-only-injected mice, although this was not observed with the anti-A. actinomycetemcomitans antibody response. As previously mentioned, Choi et al. (2000) have shown that immunization with F. nucleatum followed by challenge with P. gingivalis leads to a polarized Th2 response, whereas immunization with P. gingivalis alone leads to a Th1 response. Gemmell et al. (1998) have previously shown a predominant splenic Th1 response with high percentages of IFN-γ-positive T-cells following immunization with a high dose of P. gingivalis outer membrane antigens. These studies again highlight the complex, often synergistic, responses (Ebersole et al., 1991) with co-infection, which may have relevance to the multibacterial infection of human periodontal disease.
The influence of T-cells on periodontal bone destruction induced by endotoxin was reported in a study showing that repeated injections of Escherichia coli endotoxin into the gingiva of mice mandibles increased bone resorption in normal as well as T-cell reconstituted nude mice compared with nude mice (Ukai et al., 1996). The effects of P. gingivalis were demonstrated by Baker et al. (1994, 1999), who infected the mouths of immunocompetent mice with P. gingivalis, resulting in increased levels of specific antibody and significant bone loss. Infected severe combined immunodeficient (SCID) mice (lacking T- and B-cells) also demonstrated bone loss, although not to the same extent as immunocompetent mice. These studies support the contention that tissue destruction and bone loss in periodontal disease are determined by the lymphocytic infiltrate. In another study, T-cells derived from the spleen or Peyer's patches of normal Fischer rats after oral inoculation with P. gingivalis were adoptively transferred into nude Fischer rats. After oral challenge with live organisms, serum and salivary responses were noted in treated nude rats compared with control rats which did not receive T-cells, indicating a T-cell-dependent response to P. gingivalis. Splenic and Peyer's patch T-cells induced different responses with higher serum IgG, especially IgG2 induced by the former cells, although salivary IgA responses were similar. Less horizontal bone loss was seen in rats adoptively transferred with spleen T-cells. IgG2 responses indicate the involvement of Th1 cells with IgA, the involvement of Th2 cells suggesting that the balance between these T-cell subsets may determine whether the reponses are protective against experimental bone loss after oral challenge to P. gingivalis (Katz and Michalek, 1998).
The role of the immune response to A. actinomycetemcomitans has also been studied in rats. Taubman et al. (1984b) first showed that immunization with A. actinomycetemcomitans induced a delayed-type hypersensitivity reaction with elevated bone loss compared with sham-immunized rats. Furthermore, athymic nude rats demonstrated more bone loss than normal littermates, although bone loss in nude rats reconstituted with thymus cells was similar to that in control rats. It was concluded that T-cells can exert protective and/or destructive effects on periodontal destruction. When cells from an A. actinomycetemcomitans-specific T-cell clone (A3) were adoptively transferred into rats followed by oral infection with A. actinomycetemcomitans, significantly elevated IgG and IgM antibodies to A. actinomycetemcomitans were found, and bone loss was significantly lower than in rats which did not receive the T-cells but were orally infected with the organisms (Yamashita et al., 1991). In this study, T-cells appeared to interfere with periodontal bone loss. Recently, gingival injection of A. actinomycetemcomitans was demonstrated to induce local bone resorption in rats after the transfer of A. actinomycetemcomitans-specific Th1 clone cells but not after the transfer of Th2 cells (Kawai et al., 2000). The clone cells were retained in the gingival tissues for up to 3 days. A. actinomycetemcomitans LPS was required for this bone resorption and for A. actinomycetemcomitans-specific IgG2 production. LPS also elicited the expression of B7-1 and B7-2 molecules on gingival macrophages. The administration of CTLA4Ig, which is a functional antagonist of CD28/B7 binding, abrogated the bone resorption induced by Th1 clone cells after challenge with A. actinomycetemcomitans and LPS. The results suggested that local antigen-specific activation of Th1-type cells by B7 co-stimulation may trigger inflammatory bone resorption. The relevance of these findings to human periodontal disease, however, remains to be determined.
(VI) Cell-based Therapy in Periodontal Disease—Dendritic Cell Vaccines?
If the hypothesis that Th1 cells are associated with the stable lesion and Th2 cells are associated with the progressive lesion is true, it would follow that therapies aimed at enhancing a Th1 profile may have some role in the treatment of advanced or even refractory periodontal disease. In this context, therefore, control of the Th1/Th2 profile would be fundamental. APC differ in their abilities to induce activation of different T-cell subsets and hence the cytokine profiles induced (Sundstrom and Ansari, 1995). Dendritic cells are professional APC which have an extraordinary capacity for initiating primary and secondary T-cell responses (Steinman, 1991). Immature dendritic cells originate in the bone marrow and migrate to peripheral non-lymphoid tissues. Dendritic cells process antigen and can also recruit leukocytes to the site of inflammation by the production of chemokines, inflammatory cytokines, and interferons. In the presence of inflammatory signals such as LPS, IL-1, TNF-α, they differentiate, and a switch in chemokine receptor expression allows them to migrate from mucosal surfaces as veiled cells via the afferent lymph to the draining lymph nodes. During this process, dendritic cells down-regulate their ability to phagocytose and process antigen and up-regulate MHC class I and II molecules and the co-stimulatory molecules CD80, CD86, and CD40 and present antigen to T-cells in the T-cell areas (Pettit and Thomas, 1999; Rescigno et al., 1999). Activated T-cells in the lymph nodes may then activate B-cells. Follicular dendritic cells in the germinal follicles can stimulate memory B-cells which in turn stimulate further T-cell activity. Memory T-cells travel to the site of inflammation and can be activated at the peripheral sites. Memory T-cells can also travel via the afferent lymph back to the lymph nodes, and B-cells can carry antigen into the nodes from the circulation (Knight and Stagg, 1993).
T-cells in the gingival tissues are primed/memory cells (Gemmell et al., 1992; Yamazaki et al., 1993), and in the development of periodontal disease, plaque bacterial antigens are most likely initially presented to T-cells in the regional lymph nodes, after which sensitized T-cells home back to the gingival tissues, where they are further activated (Seymour et al., 1988). Higher numbers of CD1+ Langerhans cells have been demonstrated in the epithelium of marginal gingivitis and adult periodontitis than in healthy tissues (Meng and Zheng, 1990), and increasing numbers were shown in a 21-day experimental gingivitis study (Seymour et al., 1988). In this study, adenosine triphosphatase+ cells with a morphology typical of interdigitating dendritic cells were also demonstrated in the perivascular spaces in close contact with lymphocytes, although approximately four times as many acid phosphatase+ phagocytic macrophages were present within the lesions as well as in the subepithelial zones. Increasing numbers of B-cells express an increasingly activated phenotype in the progression from gingivitis to periodontitis (Gemmell and Seymour, 1991), while macrophage numbers do not increase in advanced periodontitis compared with minimally inflamed tissues (Chapple et al., 1998). The roles of dendritic cells, B-cells, and macrophages as APC in periodontitis have not yet been elucidated. However, unlike B-cells, macrophages and dendritic cells both bind antigen by relatively non-specific mechanisms. Macrophages and dendritic cells may therefore provide signals that initially activate T-cells, while B-cell antigen presentation may allow for further activation and clonal expansion of these already-activated cells.
Non-professional APC may initiate secondary immune responses after the induction of MHC class II molecules by cytokines such as IFN-γ (Nickoloff and Turka, 1994). Gingival keratinocytes contribute to periodontal disease progression by the secretion of several pro-inflammatory cytokines, including IL-1, IL-6, and IL-8, and the expression of adhesion molecules, which aid in the influx of leukocytes into the gingival sulcus. Epithelial tissues act as physical barriers to the outside environment; however, unlike normal plaque flora, the periodontopathogens P. gingivalis and A. actinomycetemcomitans can disrupt the gingival epithelial barrier by adhering to and invading epithelial cells and even the connective tissues in diseased sites (Sandros et al., 1993; Fives-Taylor et al., 1995), highlighting the possibility of antigen presentation of P. gingivalis and A. actinomycetemcomitans antigens by gingival keratinocytes (Saglie et al., 1988). Keratinocytes function as accessory cells by activating T-cells via the TCR/CD3 complex, and this involves signals by the adhesion molecules ICAM-1 (CD54) on the keratinocyte and LFA-1 (CD11a) on the T-cell (Nickoloff et al., 1993). Approximately 50% of cells in the infiltrates of periodontal lesions have been shown to be LFA-1+ while keratinocytes in inflamed gingiva express HLA-DR, ICAM-1, and LECAM-1 (CD62L) (Gemmell et al., 1994). As well as keratinocytes, other resident cells such as activated endothelial cells and fibroblasts may also play a role in antigen presentation in periodontitis lesions.
Suchett-Kaye et al. (1998) have suggested that T-cells in the progressive lesion may respond to a wider range of APC, including gingival keratinocytes, leading to dysregulation of the T-cell response, particularly cell-mediated immunity, as well as favoring non-protective polyclonal B-cell responses. In the study alluded to earlier, peripheral blood mononuclear cells were used as APC to present P. gingivalis antigens to P. gingivalis-specific T-cells, resulting in highly varied profiles, due possibly to presentation by dendritic cells and/or monocytes and/or B-cells (Gemmell et al., 1999). While these results were inconclusive, we have recently been able to demonstrate that when these same lines were presented with P. gingivalis antigens by autologous EBV-transformed B-cells (LCL), the cytokine profiles were predominantly IL-4+, with lower numbers of IFN-γ+ T-cells and very few IL-10+ T-cells, and these profiles were constant for every T-cell line. These results strongly suggest a central role for APC in periodontal disease.
While the function of different APC in periodontitis is mostly purely speculative, Cutler et al. (1999) have recently put forward a hypothesis on dendritic cells and P. gingivalis based on their observations of in vivo and in vitro events. P. gingivalis was identified in the epithelium and connective tissues of gingival sections from periodontitis subjects, and double immunofluorescence showed an association between P. gingivalis and immature CD1a+ Langerhans cells in the epithelium. Immature dendritic cells appeared to be limited to the epithelium, whereas mature dendritic cells were restricted to the connective tissue. It was hypothesized that immature dendritic cells could be exposed to P. gingivalis, resulting in their activation/maturation and movement into the connective tissue. In vitro evidence that dendritic cells internalize P. gingivalis was demonstrated. As well, maturation of the dendritic cells was shown by increased expression of co-stimulatory molecules, CD83 and HLA-DR. Sensitized dendritic cells were also able to stimulate T-cells to proliferate in a dose-dependent manner. P. gingivalis sensitization of dendritic cells was further hypothesized to occur in advanced gingivitis or early active periodontitis, followed by the homing of P. gingivalis-specific effector T-cells with persistent P. gingivalis infection in severe periodontitis.
In the future, appropriate APC may be used to modulate the immune response to induce protective responses in susceptible people. The use of dendritic cells as a powerful tool for manipulating the immune system is currently being examined in several areas. In particular, dendritic cells have been shown to be potent immunotherapeutic agents when pulsed with tumor-associated antigens, and trials based on their use as an effective anti-cancer treatment (Hart, 1997) are currently being conducted. Whether dendritic cells pulsed with protective antigens of periodontopathic bacteria will be an appropriate form of treatment has yet to be determined.
(VII) Conclusion
This review, as part of the "Controversy" series, has highlighted several controversial issues related to our current knowledge of the pathogenesis of periodontal disease. The review is intended to be provocative, to stimulate debate, and, importantly, to stimulate further research. In a true Popperian sense, science advances by the formation of models or hypotheses and by experiments being designed to disprove these hypotheses. In this context, if this review stimulates further research, it will have achieved its aim.
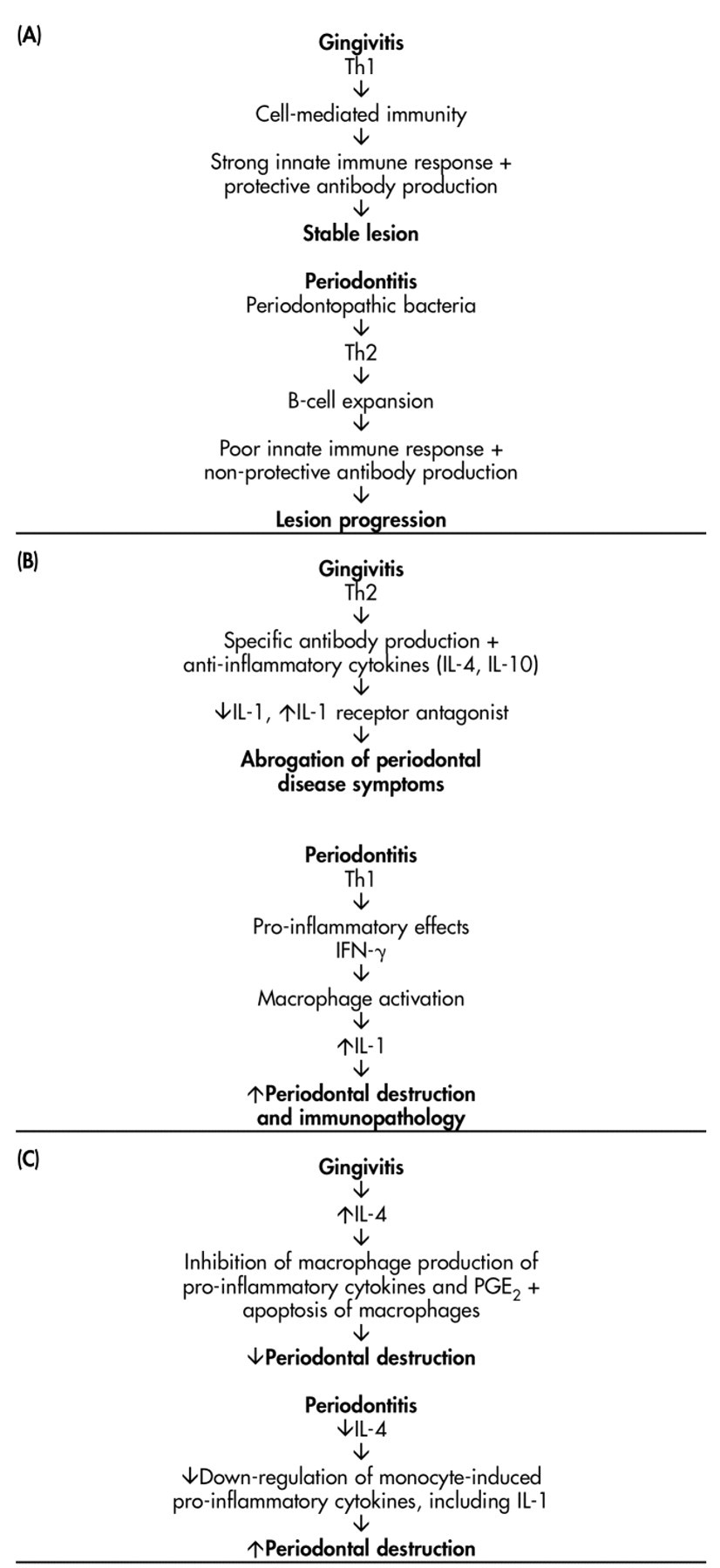
Models for the roles of Th1 and Th2 cytokines in periodontal disease progression as proposed by