
Abstract
Melanoma is a deadly disease at late metastatic stage, and early diagnosis and accurate staging remain the key aspects for managing melanoma. The melanocortin 1 receptor (MC1 R) is overexpressed in primary and metastatic melanomas, and its endogenous ligand, the α-melanocyte-stimulating hormone (αMSH), has been extensively studied for the development of MC1 R-targeted molecular imaging and therapy of melanoma. Natural αMSH is not well suited for this purpose due to low stability in vivo. Unnatural amino acid substitutions substantially stabilized the peptide, while cyclization via lactam bridge and metal coordination further improved binding affinity and stability. In this study, we summarized the development and the in vitro and in vivo characteristics of the radiolabeled αMSH analogues, including 99mTc-, 111In-, 67 Ga-, or 125I-labeled αMSH analogues for imaging with single-photon emission computed tomography; 68Ga-, 64Cu-, or 18F-labeled αMSH analogues for imaging with positron emission tomography; and 188Re-, 177Lu-, 90Y-, or 212Pb-labeled αMSH analogues for radionuclide therapy. These radiolabeled αMSH analogues showed promising results with high tumor uptake and rapid normal tissue activity clearance in the preclinical model of B16F1 and B16F10 mouse melanomas. These results highlight the potential of using radiolabeled αMSH analogues in clinical applications for molecular imaging and radionuclide therapy of melanoma.
Introduction
It is estimated that 87 110 new melanoma cases and 9 730 deaths will occur in the United States in 2017, 1 which is an average of over 1 patient death per hour. The incident rate of melanoma has been steadily increasing over the past 40 years. Melanoma accounts for about only 1% of all skin cancers but causes the majority of skin cancer deaths. Late-stage metastatic melanoma is particular deadly, with a 5-year survival rate of 34% even with the advent of new treatments such as immune checkpoint inhibitors. 2 Therefore, early diagnosis and accurate staging remain key aspects in managing melanoma.
Cancer imaging with 2-[18F]fluorodeoxyglucose ([18F]FDG) using positron emission tomography (PET) is commonly used for melanoma staging and restaging. 2-[18F]fluorodeoxyglucose can be transported into cells by glucose transporters and phosphorylated during glycolysis. The upregulation of glucose transporters and increased metabolic rate in tumor cells lead to higher signal and therefore detection in PET scans. The sensitivity and specificity of [18F]FDG PET for staging cutaneous malignant melanoma are estimated to be 83% and 85%, respectively. 3 However, the detection rate can be as low as 23% when the metastatic lesions are small (≤0.5 cm). 4 Moreover, since [18F]FDG does not target tumor-specific antigens, limited sensitivity was shown in detecting early nodal metastases and liver metastases in uveal melanoma. 5,6 Molecular probes targeting cancer-specific receptors could offer higher sensitivity and specificity for the detection of small metastatic lesions. The best example is the successful development of the radiolabeled somatostatin analogues targeting the somatostatin 2 receptor for diagnostic imaging and radionuclide therapy of neuroendocrine tumors. 7 More recently, 2-(3-(1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl)-amino]-pentyl)-ureido)-pentanedioic acid ([18F]DCFPyL) was developed and successfully used to target the prostate-specific membrane antigen (PSMA) for prostate cancer imaging 8 and showed promising results in detecting primary and metastatic tumors in patients. 9
The melanocortin 1 receptor (MC1 R) is one of the most-targeted melanoma antigens. Melanocortin 1 receptor belongs to the melanocortin family of G protein-coupled receptors, which consists of 5 receptor subtypes, that is, MC1 R to MC5 R. Distinct functions and tissue distribution of these receptors have been revealed, with MC1 R specialized in regulating skin and hair color. 10 It has been shown that MC1 R is expressed in nearly all primary and metastatic melanomas (n = 26) 11 and 95% of uveal melanoma. 12 Combined with low expression in normal tissue, MC1 R is an attractive receptor for molecular-targeted imaging and radionuclide therapy of melanoma. α-Melanocyte-stimulating hormone (αMSH), a tridecapeptide, is an endogenous ligand to the melanocortin family of receptors, with subnanomolar binding affinity to MC1 R. 13 α-Melanocyte-stimulating hormone also binds to MC3 R, MC4 R, and MC5 R with lower affinity at 31.5, 900, and 7 160 nmol/L, respectively. 13 Another MC1 R-targeted compound, MC1 RL, has recently been developed and showed its potential for melanoma imaging. 14 Besides MC1 R, melanin produced by melanocytes has also been targeted for melanoma imaging and therapy with benzamide-based small molecules 15 –19 and peptides. 20 –22
α-Melanocyte-Stimulating Hormone Analogues
An extensive effort has been made to develop αMSH analogues to improve plasma stability while maintaining excellent binding affinity to MC1 R for the purpose of imaging and radionuclide therapy of melanoma. Notable αMSH analogues are shown in Figure 1. α-Melanocyte-stimulating hormone, Ac-Ser1-Tyr2-Ser3-Met4-Glu5-His6-Phe7-Arg8-Trp9-Gly10-Lys11-Pro12-Val13-NH2, like many peptides, is prone to proteolytic degradation in vitro and in vivo. In 1980, Hadley and colleagues showed that with unnatural amino acid substitutions at Nle4 and D-Phe7, prolonged biological activity compared to native αMSH was observed, suggesting that this analogue was resistant to enzymatic degradation. 23 The resulting (Nle4, D-Phe7)αMSH, also known as NDPMSH (Figure 1), was shown to be a highly potent nonselective ligand of the melanocortin receptors with binding affinity (K i) to MC1 R, MC3 R, MC4 R, and MC5 R at 0.109, 0.469, 2.93, and 5.50 nmol/L, respectively. 13 In the early 1990s, Bard and colleagues made the first attempts to radiolabel αMSH by conjugating a chelator, diethylenetriamine pentaacetic acid (DTPA), to 2 molecules of αMSH, 24 or to 1 or 2 molecules of NDPMSH, 25 and showed tumor activity accumulation of 2.70% ± 0.24% injected dose per gram of tissue (%ID/g) at 24-hour post injection (p.i.) with good tumor-to-normal organ contrast in mice bearing cloudman S91 mouse melanoma, which demonstrated the feasibility of using αMSH analogues to target malignant melanoma.

αMSH analogues designed for the purpose of imaging and radionuclide therapy of melanoma. The minimum binding sequence is indicated by underline. Amino acid substitutions on the αMSH sequence are highlighted in bold. αMSH denotes alpha-melanocyte-stimulating hormone.
In order to identify the minimal functional sequence of αMSH, a series of truncation studies were performed by Hadley and colleagues to evaluate the potency of αMSH fragments using the classic frog and lizard skin assays. 26,27 Ac-His6-Phe7-Arg8-Trp9-NH2 was determined to be the minimal sequence for binding and biological activity. Similarly, the tripeptide, Ac-D-Phe7-Arg8-Trp9-NH2, of NDPMSH exhibited sustained activity. 28 However, with the addition of His, 6 over a 100-fold increase in potency was observed. 29 Therefore, Ac-His6-(D)-Phe7-Arg8-Trp9-NH2 served as critical building blocks for designing truncated linear and cyclized αMSH analogues. Notably, Froidevaux and colleagues designed a truncated linear NDPMSH analogue, [Ac-Nle4,Asp5,D-Phe7]-αMSH4–11 (NAPamide, Figure 1 and 2A), which displayed excellent binding affinity (half maximal inhibitory concentration [IC50]) of 0.27 nmol/L to MC1 R, radiolabeled with various isotopes and successfully used for targeting melanoma in vivo. 30

Chemical structure of αMSH analogues. (A) Linear αMSH analogue, NDPamide; (B) lactam bridge-based cyclized αMSH analogue, Nle-CycMSHhex (MTII); (C) metal coordination-based cyclized αMSH analogue, ReCCMSH; and (D) examples of linkers and chelators that enable radiolabeling. αMSH denotes alpha-melanocyte-stimulating hormone.
Cyclized αMSH analogues were also designed and represent the most promising agents for imaging and radionuclide therapy of melanoma. Cyclization of αMSH can be achieved by side chain to side chain disulfide bond formation, for example, Ac-[Cys4,Cys10] αMSH, which was introduced by Hadley and colleagues in 1982 and showed over 10 000 times more potent than the native αMSH with the frog skin assay. 31 In 1989, based on structural analysis of αMSH via molecular dynamics, an alternative cyclization strategy using lactam bridge formation was successfully developed, 32 with the general sequence of Ac-Nle4-cyclo[Xxx5,D-Phe7,Yyy10]-αMSH4-10-NH2 or Ac-Nle4-cyclo[Xxx5,D-Phe7,Yyy10, Gly11]-αMSH4-13-NH2, where Xxx = Glu or Asp, and Yyy = Lys, Orn, Dab or Dpr. Lactam bridge cyclization was achieved with the side chains of the Xxx and Yyy residues. One notable analogue, Ac-Nle4-cyclo[Asp5-His-D-Phe7-Arg-Trp-Lys10]-NH2 (Nle-CycMSHhex, or melanotan II, Figure 1 and 2B), showed 90 times increased potency compared to the native αMSH using the lizard skin assays and has been extensively modified and employed for melanoma imaging. More recently, Quinn and colleagues introduced another cyclization strategy utilizing transitional metal coordination with Re or Tc for αMSH in 1998, 33 for example, Re[Cys3,4,10, D-Phe7]-MSH3-13 (ReCCMSH, Figure 1 and 2C). Incorporation of the metal ion resulted in chemically stable and biologically active αMSH analogues. Also, radioactive 188Re or 99mTc can be directly labeled to the compounds without the need for a bifunctional chelator. These cyclized αMSH analogues have shown prolonged binding, biological activities, and higher enzymatic stability compared to the linear counterparts. 34 This is due to the rigidity in the secondary structures of the cyclic peptides, which proved to be a better fit to the receptor-binding pocket. Coupled with optional linkers and chelators (Figure 2D), many αMSH analogues have shown high tumor uptake and retention in preclinical studies.
In 2008 and 2010, 4 separate review articles have summarized the early development of radiolabeled αMSH analogues targeting MC1 R for molecular imaging and therapy of melanoma. 35 –38 In this study, we focus on the recent advances in the linear and cyclized αMSH analogues that have been labeled with radiometals or radiohalogens and evaluated their potential for melanoma imaging and radionuclide therapy. Notable radiolabeled αMSH analogues, their binding affinity to MC1 R, and biodistribution characteristics in preclinical animal models of melanoma are summarized in Table 1 for imaging with single-photon emission computed tomography (SPECT), Table 2 for imaging with PET, and Table 3 for radionuclide therapy of melanoma.
Binding Affinity and Biodistribution Data for Radiolabeled αMSH Analogues for SPECT Imaging of Melanoma.a

Abbreviations: IC50, half maximal inhibitory concentration; ID, injection dose; N/A, not applicable; p.i., post injection; αMSH denotes alpha-melanocyte-stimulating hormone.
a Table is sorted by the average tumor uptake.
b K i value.
c K d value.
Binding Affinity and Biodistribution Data for Radiolabeled αMSH Analogues for PET Imaging of Melanoma.a

Abbreviations: IC50, half maximal inhibitory concentration; ID, injection dose; N/A, not applicable; p.i., post injection; αMSH denotes alpha-melanocyte-stimulating hormone.
a Table is sorted by the average tumor uptake.
Binding Affinity and Biodistribution Data for Radiolabeled αMSH Analogues Designed for Radionuclide Therapy of Melanoma.
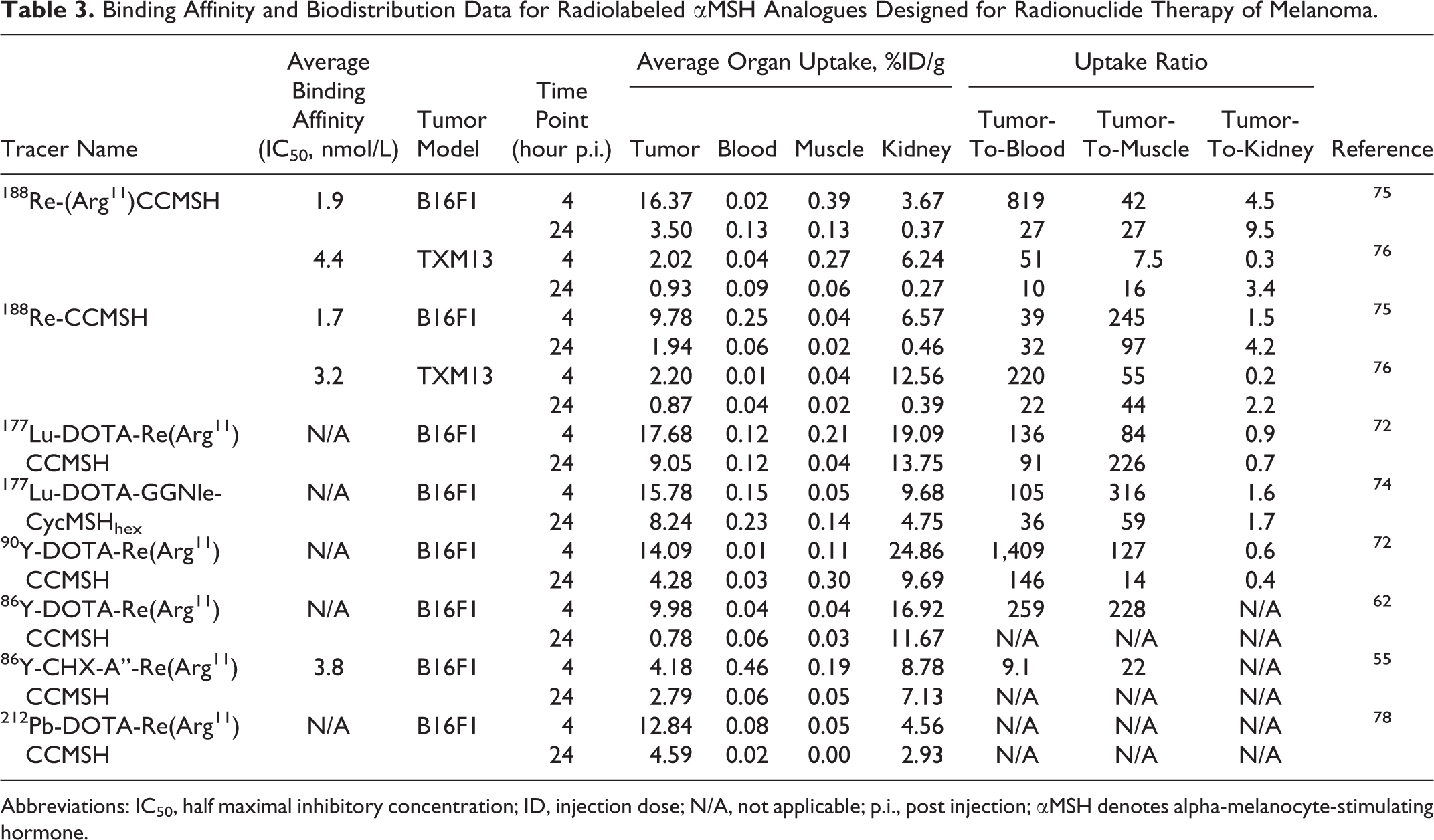
Abbreviations: IC50, half maximal inhibitory concentration; ID, injection dose; N/A, not applicable; p.i., post injection; αMSH denotes alpha-melanocyte-stimulating hormone.
α-Melanocyte-Stimulating Hormone Analogues for SPECT Imaging
Single-photo emission computed tomography is one of the most widely used imaging modalities in nuclear medicine, thanks to the availability and affordability of the γ scanning equipment and SPECT isotopes. α-Melanocyte-stimulating hormone analogues that were radiolabeled with 99mTc, 111In, 67Ga, or 125I and evaluated for melanoma imaging with SPECT are summarized in Table 1.
99mTc
99mTc is an ideal isotope for SPECT imaging due to its favorable half-life of 6.0 hours, and it decays mainly through gamma emission (140 keV) and can be readily produced from 99Mo–99mTc commercial generators. The relatively short half-life and moderate energy translates into low radiation dose and combined with the cost-effectiveness of the isotope, 99mTc has become the most commonly adopted radioisotope for medical imaging.
Thirty-eight 99mTc-labeled αMSH analogues have been developed and evaluated in preclinical settings so far (Table 1). Early development was based on the linear NDPMSH structure, and radiolabeling with 99mTc was performed via the addition of tetrafluorophenyl mercapto-acetylglycylglycyl-γ-aminobutyrate on the Lys residue or a Cys-Gly-Cys-Gly peptide chelating moiety at the N-terminus. 39 Low tumor uptake (<1%ID/g) was observed for both 99mTc-labeled αMSH analogues at 4-hour p.i. in mice bearing B16F1 mouse melanoma. 99mTc can also be radiolabeled in the form of 99mTc(CO)3, and coupled to a chelating moiety containing a pyrazolyl-diamine (pz) backbone. This was applied to a truncated NDPMSH, where the pz moiety was attached to the amino group of the lysine residue, and the resulting (Ac-Nle4,Asp5,D-Phe7,Lys11[pz-99mTc(CO)3])-αMSH4–11 showed moderate tumor uptake at 4.24%ID/g ± 0.94%ID/g at 4-hour p.i. in mice bearing B16F1 mouse melanoma. 40
Metal coordination cyclization leads to more successful αMSH analogues for melanoma imaging. Quinn and colleagues designed 99mTc[Cys3,4,10, DPhe7]αMSH3–13 (99mTcCCMSH), where Tc was stabilized by the 3 sulfhydryls in Cys3,4,10 and the amide nitrogen in Cys433. The cyclization through metal coordination was structurally stable and resistant to proteolytic degradation. Good tumor uptake was observed at 9.51%ID/g ± 1.97%ID/g at 4-hour p.i. in mice bearing B16F1 mouse melanoma, despite high kidney accumulation at 14.60%ID/g ± 1.88%ID/g. 33 With Arg11 substitution, tumor uptake increased to 11.16%ID/g ± 1.77%ID/g and kidney uptake decreased to 5.53%ID/g ± 1.17%ID/g in the same model. 41 Many modifications were made to improve the melanoma targeting potential of the 99mTc-(Arg11)CCMSH analogues (Table 1). Improvement in tumor targeting was observed as the expense of extremely high kidney activity accumulation, in some cases, over 100%ID/g average kidney uptake. 42,43 Recently, CCMSH analogues were also employed to image human M21 melanoma, where moderate tumor uptake of 2.35%ID/g ± 0.12%ID/g and 1.71%ID/g ± 0.25%ID/g at 2-hour p.i. was obtained for 99mTc-RGD-Aoc-(Arg11)CCMSH and 99mTc-RGD-PEG2-(Arg11)CCMSH, respectively. 44 The difference in tumor uptake compared to the mouse melanoma model was due to the lower MC1 R density in the human melanoma model.
The most successful class of 99mTc-labeled αMSH analogues was designed based on the cyclized core structure of Nle-CycMSHhex. Miao and colleagues developed 99mTc(ethylenediaminediacetic acid [EDDA])- hydrazinonicotinamide (HYNIC)-GGNle-CycMSHhex, where 99mTc was radiolabeled in the presence of EDDA and coupled to the bifunctional chelator HYNIC, which was covalently attached to the Nle-CycMSHhex through a GlyGly amino acid linker. 45 This analogue showed high tumor uptake of 13.23%ID/g ± 2.35%ID/g at 4-hour p.i. in mice bearing B16F1 mouse melanoma. 45 The same study also examined the impact of 2 other chelating groups, that is, mercaptoacetyltriglycine, and Ac-Cys-Gly-Gly-Gly, on the in vivo characteristics of the 99mTc-labeled αMSH analogues, which showed lower tumor uptake of 4.64%ID/g ± 1.06%ID/g and 9.76%ID/g ± 0.51%ID/g at 2-hour p.i., respectively. 45 Although binding affinities of these 3 analogues to MC1 R were comparable, ranging from 0.6 to 1.2 nmol/L (IC50), the use of the different chelating groups showed a clear impact on the melanoma-targeting potential. In addition, employing the 99mTc(CO)3-pz labeling strategy, 99mTc(CO)3-pz-βAla-Nle-CycMSHhex was developed and yielded a tumor uptake of 11.31%ID/g ± 1.83%ID/g at 4-hour p.i. in mice bearing B16F1 mouse melanoma. 46 In comparison with the low tumor uptake (0.99%ID/g ± 0.08%ID/g) using a linear analogue, 99mTc(CO)3-Pz-βAla-Nle-Asp-DPhe-Arg-Trp-Lys-NH2, in the same animal model, the advantage of using the cyclized Nle-CycMSHhex was highlighted. 46 Modifications to the Pz chelating group were attempted; however, lower tumor uptake was observed with average ranging from 4.15%ID/g to 9.51%ID/g under the same condition. 47
Recently, Miao and colleagues further optimized the Nle-CycMSHhex analogue by the introduction of an 8-aminooctanoic acid linker (Aoc), that is, 99mTc(EDDA)-HYNIC-AocNle-CycMSHhex. Single-photon emission computed tomography imaging and biodistribution studies showed a high tumor uptake of 22.17%ID/g ± 5.93%ID/g with a moderate kidney uptake of 6.10%ID/g ± 0.72%ID/g at 4-hour p.i. in mice bearing B16F1 mouse melanoma (Figure 3). 48 Radioactivity was rapidly cleared from the normal tissue and produced excellent tumor-to-normal tissue contrast, with tumor-to-blood and tumor-to-muscle uptake ratios at 75 and 503, respectively. Besides the Aoc linker, αMSH analogues with PEG2, GlyGlyGly, and GlySerGly linker systems were also evaluated in the same study. 48 All of these Nle-CycMSHhex-based peptides exhibited subnanomolar binding affinity to MC1 R. However, the linkers showed a significant impact on radioactivity accumulation of the tumors. For instance, with the GlySerGly linker, tumor uptake of 7.41%ID/g ± 4.26%ID/g was obtained at 2-hour p.i., which was only approximately one-third of the tumor uptake compared to the Aoc linker analogue. The 99mTc(EDDA)-HYNIC-AocNle-CycMSHhex was also evaluated in a human M21 melanoma model. Due to the low expression level MC1 R compared to the mouse model, a moderate tumor uptake of 3.36%ID/g ± 1.48%ID/g at 4-hour p.i. was acquired. 49
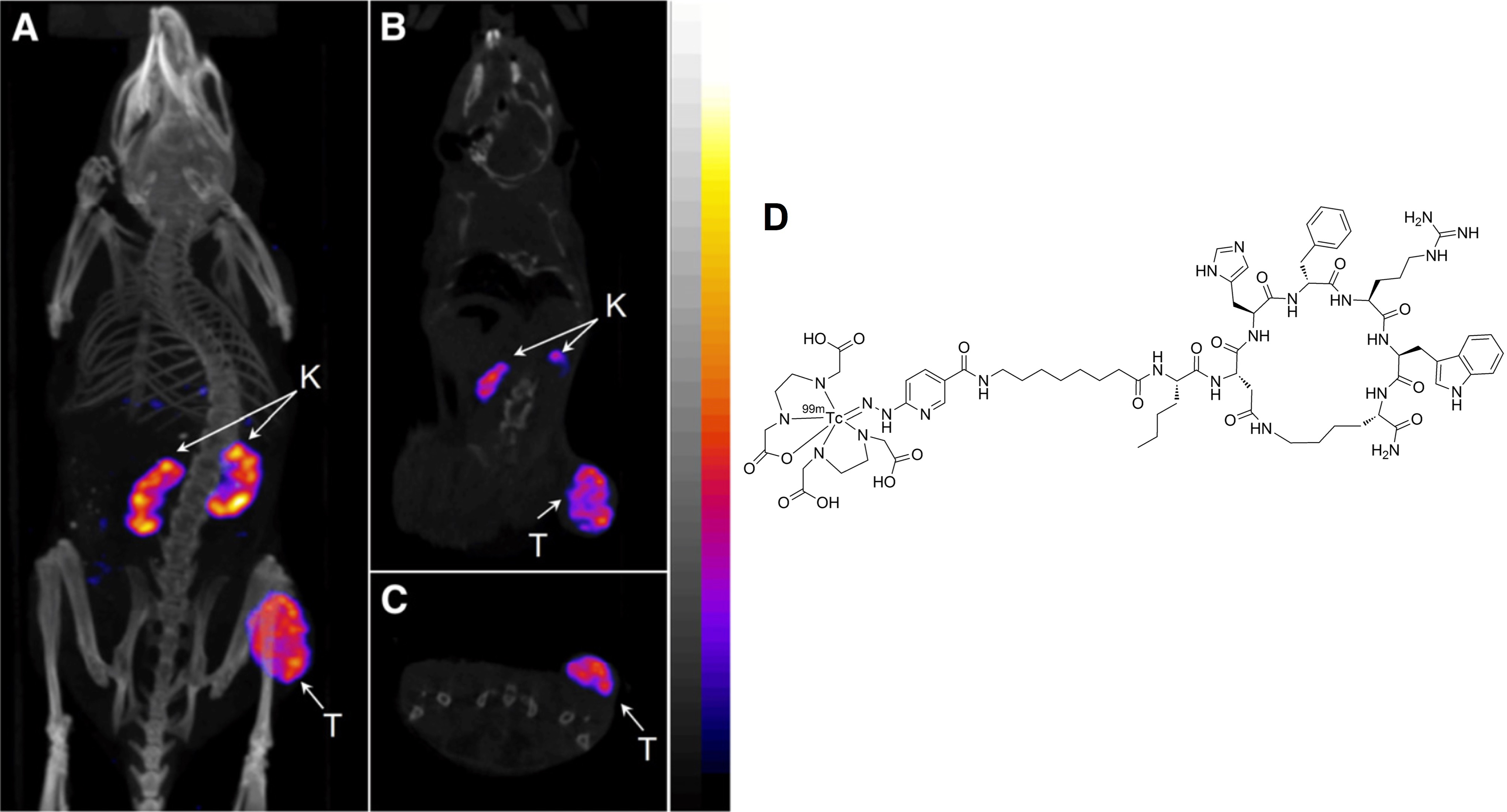
Representative SPECT/CT images of 99mTc(EDDA)-HYNIC-AocNle-CycMSHhex in C57 mice bearing B16F1 melanoma tumors at 2 hours postinjection. (A) Whole body, (B) coronal, and (C) transversal images. (D) Chemical structure of 99mTc(EDDA)-HYNIC-AocNle-CycMSHhex. This research was originally published in JNM48 and adapted with permission by the Society of Nuclear Medicine and Molecular Imaging, Inc. CT denotes computed tomography; K, kidney; SPECT, single-photon emission computed tomography; T, tumor.
111In
111In is also a popular isotope for SPECT imaging due to its manageable 2.8 day half-life and γ emissions at 171 and 245 keV. 111In can be effectively radiolabeled with chelating agents, such as DTPA and 1,4,7,10- tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA). Linear αMSH analogues labeled with 111In showed limited success due to the lower binding affinity and in vivo stability (Table 1). Nonetheless, 111In-DOTA-NAPamide showed promising results with good tumor uptake at 7.56%ID/g ± 0.51%ID/g and moderate kidney uptake at 5.06%ID/g ± 0.32%ID/g at 4-hour p.i. in mice bearing B16F1 tumors. 30
Similar to the 99mTc-labeled compounds, the most successful αMSH analogues are based on the lactam bridge and metal coordination cyclization. 111In-labeled lactam bridge cyclization was first evaluated on the αMSH analogues between Lys and Asp to form a long 12 amino acid ring, named CycMSH. 50 Using this strategy, Miao and colleagues showed that the 111In-DOTA-CycMSH and 111In-DOTA-GlyGlu-CycMSH produced good tumor uptake of 7.54%ID/g ± 0.70%ID/g and 7.40%ID/g ± 0.43%ID/g at 4-hour p.i. in B16F1 melanoma-bearing mice, respectively. 50 High kidney uptake was observed at 21.69%ID/g ± 0.34%ID/g and 12.13%ID/g ± 1.17%ID/g, respectively. Using a similar approach, Nle-CycMSHhex, which contains only 6 amino acids in the ring structure using the same Lys and Asp lactam bridge, was evaluated for melanoma targeting. 111In-DOTA-Nle-CycMSHhex showed a high tumor uptake at 17.01%ID/g ± 2.54%ID/g and a moderate kidney uptake at 9.99%ID/g ± 1.39%ID/g at 4-hour p.i. in B16F1 melanoma-bearing mice. 51 More recently, coupled with a GlyGly (GG) linker, 111In-DOTA-GGNle-CycMSHhex exhibited improved tumor take at 18.6%ID/g ± 3.56%ID/g and reduced kidney uptake at 6.82%ID/g ± 1.19%ID/g in the same condition. 52 Rapid normal tissue activity clearance lead to extremely high tumor to background contrast, where tumor-to-blood and tumor-to-muscle ratios reached 1 860 and 930, respectively. The same study also evaluated GlyGlu as the linker instead of GlyGly, which showed a much lower tumor uptake at 5.3%ID/g ± 2.84%ID/g at 4-hour p.i. as well as much lower tumor to background contrast in the same model, suggesting the negatively charged glutamic acid negatively impacted the melanoma targeting capability. 52
Cyclization with metal coordination has also seen successful development. The in vivo distribution of a metal-coordinated 111In-DOTA-ReCCMSH was compared to a nonmetalated linear analogue, 111In-DOTA-CCMSH; a disulfide bond cyclized analogue, 111In-DOTA-CMSH; and a linear analogue, 111In-DOTA-NDPMSH by Quinn and colleagues. 53 Thanks to the improved in vivo stability that metal coordination provided, 111In-DOTA-ReCCMSH showed the highest tumor uptake at 9.49%ID/g ± 0.90%ID/g and the highest tumor to background contrast at 4-hour p.i. in mice bearing B16F1 tumors. 53 Both cyclized analogues showed higher tumor uptake to the linear counterparts, demonstrating the benefits of peptide cyclization for the αMSH analogues. Interestingly, the linear NDPMSH analogue cleared slowly from muscle and showed a relatively high muscle uptake of 3.32%ID/g ± 0.51%ID/g at 4-hour p.i. in the same model. 53 In a separate study, Re-cyclized analogues were compared, including 111In-DOTA-ReCCMSH, 111In-DOTA-(Arg11)ReCCMSH, 111In-DOTA-ReCCMSH-OH, 111In-DOTA-ReCCMSH-Asp-OH, and Ac-Lys(111In-DOTA)-ReCCMSH, among which 111In-DOTA-(Arg11)ReCCMSH showed a much improved tumor uptake at 17.41%ID/g ± 5.61%ID/g and a moderate kidney uptake at 7.37%ID/g ± 1.13%ID/g at 4-hour p.i. in mice bearing B16F1 tumors, 54 making it one of the most successful 111In-labeled αMSH analogues. An attempt to change the DOTA chelator to N-(2-aminoethyl)-trans-1,2-diaminocyclohexane-N,N′,N″-pentaacetic acid (CHX-A″) did not yield favorable results, as only moderate tumor uptake was observed for 111In-CHX-A″-(Arg11)ReCCMSH at 3.87%ID/g ± 1.03%ID/g at 4-hour p.i. in the same tumor model. 55
67Ga
67Ga has a 3.3-day half-life with γ emissions at 93, 184, 300, and 393 keV and is generally produced by cyclotrons. Due to the popularity of 99mTc and 111In, and the emergence of isotopes for PET imaging, such as 68Ga and 18F, 67Ga has seen less and less usage over the past decade. Similar to 111In, 67Ga can be radiolabeled effectively with a DOTA chelator. 67Ga-labeled DOTA-NAPamide also produced good in vivo results as a linear αMSH analogue, that is, tumor uptake at 9.43%ID/g ± 1.06%ID/g and kidney uptake at 3.98%ID/g ± 0.10%ID/g at 4-hour p.i. in mice bearing B16F1 tumors. 30
Using the lactam bridge cyclization of Nle-CycMSHhex and the GlyGly linker, Miao and Colleagues showed that 67Ga-DOTA-GGNle-CycMSHhex produced an excellent tumor uptake of 25.13%ID/g ± 4.13%ID/g and a moderate kidney uptake of 8.44%ID/g ± 0.11%ID/g at 4-hour p.i. in mice bearing B16F1 tumors. 56 Changing the DOTA chelator to 1,4,7-triazacyclononane-triacetic acid (NOTA), which also forms stable complex with 67Ga, yielded slightly lower tumor uptake at 18.17%ID/g ± 4.89%ID/g and kidney uptake at 7.58%ID/g ± 2.70%ID/g under the same condition. 56 Another study evaluated the lactam bridge cyclized CycMSH labeled with 67Ga, that is, 67Ga-DOTA-GlyGlu-CycMSH. 57 The 12 amino acid ring analogue generated less favorable results compared to the shorter Nle-CycMSHhex ring, where tumor uptake of 8.12%ID/g ± 0.60%ID/g and high kidney uptake of 22.6%ID/g ± 4.03%ID/g was observed at 4-hour p.i. in mice bearing B16F1 tumors.
125I
125I has seen limited usage due to in vivo dehalogenation. Ac-DLys(125I-IBA)-ReCCMSH(Arg11) and Ac-Lys(125I-IBA)-ReCCMSH(Arg11) showed high tumor uptake at 15.10%ID/g ± 1.38%ID/g and 14.94%ID/g ± 2.34%ID/g at 4-hour p.i. in mice bearing B16F1 tumors, respectively. 58 However, overall high background organ activity accumulation was also observed, which prevented successful usage in SPECT imaging.
α-Melanocyte-Stimulating Hormone Analogues for PET Imaging
Compared to SPECT, PET imaging offers better image quality, higher sensitivity, and spatial resolution. With the increasing usage of [18F]FDG, PET imaging has gain significant popularity over the past decade. Limited options are available for αMSH-based PET imaging agents targeting melanoma, in comparison to the large number of compounds described earlier for SPECT. α-Melanocyte-stimulating hormone analogues that were radiolabeled with 68Ga, 64Cu, or 18F and evaluated for melanoma imaging with PET are summarized in Table 2.
68Ga
68Ga is a positron emitter (88%) with a relatively short half-life of 67.7 minutes. 68Ga can be readily produced by a 68Ge/68Ga commercial generator, enabling on-site production without the need for a cyclotron. Like 111In and 67Ga, 68Ga can be effectively radiolabeled with a chelator, such as DOTA.
Metal coordination cyclized αMSH analogues were radiolabeled with 68Ga and showed limited success. 68Ga-DOTA-Re(Arg11)CCMSH59 and 68Ga-CHX-A″-(Arg11)ReCCMSH55 in 2 separate studies showed relatively low tumor uptake of 4.25%ID/g ± 1.41%ID/g and 2.68%ID/g ± 0.69%ID/g at 2-hour p.i. in mice bearing B16F1 tumors, respectively.
Based on the lactam bridge cyclized αMSH analogues, we recently designed 3 Nle-CycMSHhex-based peptides, namely, CCZ01047, CCZ01048, and CCZ01056, with a neutral PEG2 linker, a positively charged 4-amino-(1-carboxymethyl) piperidine (Pip) linker, and dual Pip linkers, respectively (Table 2). 60 Interestingly, with the introduction of a cationic Pip linker, 68Ga-DOTA-Pip-Nle-CycMSHhex (CCZ01048) showed excellent tumor visualization (Figure 4), with tumor uptake at 21.9%ID/g ± 4.63%ID/g and moderate kidney uptake at 5.51%ID/g ± 0.39%ID/g at 2-hour p.i. in mice bearing B16F10 melanoma. Rapid normal tissue activity clearance was observed with tumor-to-kidney uptake ratio at 4.0. The high ratio of tumor-to-kidney uptake could be particularly advantageous when swapping 68Ga with a therapeutic radioisotope, so that kidneys, the dose limiting organ, would have lower irradiation dose.

A, Representative PET/CT image for 68Ga-CCZ01048 at 2 hours postinjection in mice bearing B16F10 tumor. B, Chemical structure of 68Ga-CCZ01048. The scale bar unit is %ID/g. Tumor is indicated by an arrow. CT denotes computed tomography; ID, injected dose; PET, positron emission tomography.
64Cu
64Cu has a half-life of 12.7 hours and decays by positron emission (17.9%) and β decay (39.0%), which allow applications for PET imaging. Miao and colleagues evaluated 64Cu-labeled lactam bridge cyclized αMSH analogue, 64Cu-DOTA-GGNle-CycMSHhex and showed moderate tumor uptake of 5.25%ID/g ± 1.22%ID/g and high background normal tissue uptake, including liver (9.61%ID/g ± 1.34%ID/g), kidneys (7.45%ID/g ± 0.96%ID/g), and stomach (5.09%ID/g ± 0.69%ID/g) at 4-hour p.i. in mice bearing B16F1 melanoma. 61 This was presumed to be caused by free 64Cu leaking from the DOTA chelator. The same study reported a more suitable chelator, NOTA, and 64Cu-NOTA-GGNle-CycMSHhex had significantly higher tumor uptake of 12.71%ID/g ± 2.68%ID/g with much lower normal organ uptake, for example, kidneys at 3.53%ID/g ± 0.57%ID/g and liver at 0.75%ID/g ± 0.17%ID/g in the same tumor model. 61
Similarly, for 64Cu-labeled metal coordination cyclized αMSH analogue, 64Cu-DOTA-ReCCMSH(Arg11), a moderate tumor uptake was observed at 7.35%ID/g ± 1.47%ID/g with high background activity accumulation including liver at 7.34%ID/g ± 1.79%ID/g at 4-hour p.i. in mice bearing B16F1 melanoma. 62 In a separate study, using a more suitable chelator, 4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo(6.6.2)hexadecane (CBTE2A),64Cu-CBTE2A-ReCCMSH (Arg11) produced comparable tumor uptake at 7.37%ID/g ± 1.26%ID/g with reduced overall normal tissue uptake including liver (1.77%ID/g ± 0.12%ID/g) at 4-hour p.i. in the same animal model. 63
In addition, a 64Cu-labeled linear αMSH analogue, 64Cu-DOTA-NAPamide, showed moderate tumor uptake of 4.43%ID/g ± 0.94%ID/g at 4-hour p.i. in mice bearing B16F10 melanoma64 and was not further evaluated with other chelating groups.
18F
18F is the most widely used PET isotope due to the popularity of [18F]FDG. 18F decays 97% of the time through positron emission with 109.8 minutes of half-life. However, the preclinical data for the18F-labeled αMSH analogues have been suboptimal (Table 2). Cheng and colleagues performed 18F labeling through N-succinimidyl-4-18F-fluorobenzoate (18F-SFB) or 4-nitrophenyl-2-18F-fluoropropionate (18F-NFP) on metal coordinated cyclized ReCCMSH or linear NAPamide analogues, which produced tumor uptake <2%ID/g in mice bearing B16F10 tumors. 65 –67 Further investigations are urgently needed to develop 18F-labeled αMSH analogues for melanoma imaging with PET.
α-Melanocyte-Stimulating Hormone Analogues for Radionuclide Therapy
Peptide receptor radionuclide therapy has seen significant development over the past few decades. 68 –70 Thanks to high tumor receptor binding affinity, peptides can be used as delivery vehicles, when labeled with cytotoxic radionuclide, to specifically target and kill cancerous cells. Peptide-based compounds for radionuclide therapy have distinct advantages, since they are generally cleared rapidly from circulation and therefore low activity accumulation in normal tissues is expected, with the exception of kidneys. When coupled with a bifunctional chelator, such as DOTA, these peptides can be radiolabeled with a diagnostic imaging isotope, for example, 111In, 68Ga, and 64Cu; the same peptides can alternatively be radiolabeled with a therapeutic α or β emitter, for example, 225Ac and 177Lu. Such compounds that can be employed for both therapeutic and diagnostic purposes are commonly referred to as “theranostic” agents. Diagnostic imaging can be used to monitor disease progression and allow dosimetry estimation for personalized radionuclide therapy using the same peptide when later labeled with a therapeutic radioisotope.
α-Melanocyte-stimulating hormone analogues are suitable for radionuclide therapy, since they bind to MC1 R with subnanomolar affinity, excellent in vivo stability with the cyclized analogues as well as rapid internalization. 42,46 The most successful αMSH analogues have been radiolabeled with177Lu,188Re, 90Y, and 212Pb and were evaluated for their effectiveness in melanoma treatment (Table 3). Tumor uptake values at both 4 and 24-hour p.i. were summarized to reflect radioactivity retention at these time points.
177Lu
177Lu is a radioisotope with moderate mean β emission energy (0.57 MeV), 6.7 day half-life, and 1.5 mm maximum particle range. 71 The moderate energy and short particle range is well suited for treating small tumors and metastasis. As discussed earlier, 111In-DOTA-Re(Arg11)CCMSH is one of the most successful αMSH analogues, Miao and colleagues showed that the 177Lu-labeled counterpart, 177Lu-DOTA-Re(Arg11)CCMSH, also yielded a high tumor uptake of 17.68%ID/g ± 3.32%ID/g and high kidney uptake of 19.09%ID/g ± 2.38%ID/g at 4-hour p.i. in mice bearing B16F1 tumors. 72 In addition, the 177Lu-labeled peptide exhibited rapid cellular internalization in vitro. In a separate study, administration of a single dose of 37 MBq or 2 doses of 18.5 MBq of 177Lu-DOTA-Re(Arg11)CCMSH was evaluated in mice bearing B16F1 tumors, which showed moderate but statistically significant improvement in the mean survival time from 13.3 ± 2.3 days of control group to 15.1 ± 1.8 and 16.2 ± 3.6 days, respectively. 73 Despite the high kidney uptake, no acute renal toxicity was observed. More recently, building on the success of DOTA-GGNle-CycMSHhex analogues, Miao and colleagues showed that 177Lu-DOTA-GGNle-CycMSHhex also produced high tumor uptake of 15.78%ID/g ± 1.45%ID/g and reduced kidney uptake of 9.68%ID/g ± 1.95%ID/g at 4-hour p.i. in B16F1 tumor-bearing mice (Figure 5). 74 The effectiveness of this therapeutic agent would need to be further evaluated.

A, Representative SPECT/CT image for 177Lu-DOTA-GGNle-CycMSHhex in mice bearing B16F1 tumor at 2 hours postinjection. B, Chemical structure of 177Lu-DOTA-GGNle-CycMSHhex. Tumor is indicated by an arrow. Figure adapted with permission from Guo et al. 74 CT denotes computed tomography; SPECT, single-photon emission computed tomography.
188Re
188Re is a high-energy β emitter (2.13 MeV) with a 17.0-hour half-life and maximum particle range of 11.0 mm71 and can be produced via a 188W–188Re generator. The high energy and long particle range is applicable for treating large tumors. 188Re was directly radiolabeled to cyclized αMSH analogues via metal coordination by Quinn and colleagues, that is, 188Re-(Arg11)CCMSH and 188Re-CCMSH, which produced high tumor uptake of 16.37%ID/g ± 3.27%ID/g and 9.78%ID/g ± 2.00%ID/g at 4-hour p.i. in mice bearing B16F1 tumors, respectively. 75 Minimal background activity accumulation was observed with moderate kidney uptake at 3.67%ID/g ± 0.51%ID/g and 6.57%ID/g ± 1.08%ID/g, respectively. Amino acid coinfusion was also evaluated and helped reduce kidney activity accumulation and improved tumor-to-kidney uptake ratio by up to 46.8%. 75 Similarly, the biodistribution of 188Re-(Arg11)CCMSH and188Re-CCMSH was also evaluated in the human TXM13 melanoma model and showed moderate tumor uptake of 2.02%ID/g ± 0.27%ID/g and 2.20%ID/g ± 0.24%ID/g at 4-hour p.i., respectively. 76 This was due to the low expression level of MC1 R on human TXM13 cells compared to the mouse B16F10 or B16F1 cells. In a preclinical therapy study, administration of a single dose of 7.4 or 22.2 MBq or 2 doses of 14.8 MBq of 188Re-(Arg11)CCMSH was used to treat melanoma in mice bearing mouse B16F1 tumors, where all treatment groups showed substantial tumor growth inhibition compared to the control group. 77 The 2 × 14.8 MBq treatment resulted in moderate but significant increase in mean survival time from 9.4 ± 1.1 days of the control group to 13.3 ± 1.9 days. Kidneys were determined to be the dose-limiting organ and showed no evident radiation-induced damage. The same study also evaluated administration of a single dose of 37 or 22.2 MBq, or 2 doses of 14.8 MBq of 188Re-(Arg11)CCMSH in mice bearing human TXM13 tumors, 77 where treatment with a single dose of 37 or 22.2 MBq showed significant mean survival time increase from 39.6 ± 15.0 days of control group to 72.7 ± 18.3 and 57.6 ± 24.2, respectively. Histopathologic examinations showed no evidence of dose-related toxicity.
90Y
90Y is also a high-energy β emitter (2.28 MeV) with a half-life of 64.1 hours and maximum particle range of 12.0 mm. 71 Similar to the 177Lu-labeled counterpart, 90Y-DOTA-Re(Arg11)CCMSH showed high tumor uptake of 14.09%ID/g ± 2.73%ID/g and high kidney uptake of 24.86%ID/g ± 4.89%ID/g at 4-hour p.i. in mice bearing B16F1 tumors. 72 Also, the biodistribution of 86Y-labeled cyclized αMSH analogues were evaluated, 55,62 which could be used as guidance for in vivo behavior of the 90Y-labeled counterparts. The efficacy of the 90Y-labeled peptides would need to be evaluated in therapy studies.
212Pb
212Pb is a low-energy β emitter (between 40 and 60 keV) with a half-life of 10.6 hours and can be produced by 224Ra–212Pb generators. Interestingly, its daughter isotope, 212Bi, is a high-energy α emitter (7.80 MeV) with a short half-life of 60.6 minutes and maximum particle range of 70.0 μm 71 .The short range can be translated into 3 to 5 cells in diameter, which could minimize normal tissue damage. Due to the half-life being more than 10 times longer, 212Pb is more advantageous and produces sustained irradiation dose than using 212Bi alone. 212Pb-DOTA-Re(Arg11)CCMSH showed high tumor uptake of 12.84%ID/g ± 2.53%ID/g and moderate kidney uptake of 4.56%ID/g ± 1.27%ID/g at 4-hour p.i. in mice bearing B16F1 tumors. 78 For therapeutic effect, 7.4, 3.7, or 1.85 MBq of 212Pb-DOTA-Re(Arg11)CCMSH was administered to mice bearing B16F1 tumors; 45% and 20% of mice survived the 120-day study in the 7.4 and 3.7 MBq groups, respectively. For those who did not survive the whole study, the mean survival time was 49.8, 28.0, and 22.0 days for the 7.4, 3.7, and 1.85 MBq groups, respectively. This was a significant increase from the 14.6-day mean survival time from the control group. However, moderate kidney toxicity was observed for the 7.4 MBq dose, with 3.7 and 1.85 MBq groups showing minor kidney damage.
Perspective and Future Direction
There has been significant advancement in the development of radiolabeled αMSH analogues for MC1 R-targeted melanoma imaging and therapy in recent years. The native αMSH peptide is not suitable for this purpose due to proteolytic degradation in vitro and in vivo. With unnatural amino acid substitutions and cyclization through lactam bridge and metal coordination, 3 major classes of αMSH analogues were successfully developed, that is, NAPamide (Figure 2A), Nle-CycMSHhex (Figure 2B), and ReCCMSH (Figure 2C). Compared to the linear NAPamide, cyclized αMSH analogues have shown further improvement in vitro binding affinity at subnanomolar range as well as improved in vivo stability due to the rigid ring structure. Of the 2 classes of cyclized αMSH analogues, the Nle-CycMSHhex-based peptides showed faster normal tissue clearance in vivo, particularly lower kidney activity accumulation compared to the ReCCMSH analogues. As a result, the recently developed 99mTc(EDDA)-HYNIC-AocNle-CycMSHhex and 68Ga-DOTA-Pip-Nle-CycMSHhex represent the most promising SPECT and PET αMSH analogues to date for MC1 R-targeted imaging. Both these radiotracers have shown outstanding tumor uptake at >20%ID/g at 2- or 4-hour p.i. with a relatively low average kidney uptake at 5.5%ID/g to 6.1%ID/g in the preclinical animal model bearing mouse B16F1 or B16F10 melanoma. Excellent SPECT/CT (Figure 3) and PET/CT (Figure 4) images were generated using these αMSH analogues with high tumor-to-normal organ contrast.
Radionuclide therapy studies were performed mainly with the ReCCMSH analogues, which showed promising results in increased survival rate in mice bearing B16F1 melanoma. Since kidneys are the dose-limiting organ, the lower kidney uptake in Nle-CycMSHhex-based peptides could be translated into lower normal tissue damage and/or higher radioactivity allowed to be administered and in turn higher dose at tumor sites. Therefore, the Nle-CycMSHhex analogues would potentially be more effective in therapy experiments, and further investigation is needed.
However, even with the much improved resistance to in vivo proteolytic degradation of the cyclized αMSH analogues, tumor uptake was not sustained over longer period of time. For instance, tumor uptake of 99mTc(EDDA)-HYNIC-AocNle-CycMSHhex reduced from 22.17%ID/g ± 5.93%ID/g at 4-hour p.i. to 7.13%ID/g ± 0.99%ID/g at 24-hour p.i. in mice bearing B16F1 tumor. 48 Similar trend has also been observed with other αMSH analogues radiolabeled with various isotopes for both imaging and therapy purposes (Table 3). This indicates that further in vivo stability improvement might be required.
Moreover, the MC1 R expression level on the mouse B16F1 and B16F10 tumors is 6 to 15 times higher than human melanoma, such as M21, TXM13, and A375 M cells, which only express MC1 R from a few hundred to 5 700 copies per cell. 44,49,76 The preclinical results in human melanoma model have been suboptimal with a low tumor uptake in the range of 1.71 to 3.26%ID/g (Table 1 –3). Further optimization is still required. One critical aspect would be improvement in specific activity of the radiolabeled compounds. Low receptor counts demand high specific activity to reduce the chances of tumor binding site saturation, which would in turn increase tumor uptake in the human melanoma models.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the Canadian Institutes of Health Research (FDN-148465 and MOP-119361).