
Abstract
Thevissen K, Ny A, Copmans D, Tits J, Kamata K, Gielis E, Longin K, Sourbron J, Thergarajan P, Tan TH, Ali I, Jones NC, O’Brien TJ, Monif M, Semple BD, Germeys C, Frizzi B, Minniti V, Perrone S, Linster CL, Elia I, Van Den Bosch L, Cammue BPA, Voet A, Lagae L, de Witte P. Epilepsia. 2025. doi:10.1111/epi.18536. Online ahead of print Objective: Drug-resistant epilepsy (DRE) affects >25 million people worldwide and is often associated with neuroinflammation. Increasing evidence links deficiency or malfunctioning of the enzyme phosphoglycerate dehydrogenase (PHGDH), which converts 3-phosphoglycerate to generate serine and the neurotransmitter glycine, with (drug-resistant) epilepsy. Moreover, PHGDH, which is primarily expressed in astrocytes within the brain, has been identified as a critical enzyme in driving macrophage polarization toward an anti-inflammatory state. Hence, PHGDH activators may be beneficial for treating DRE by exhibiting both antiseizure and anti-inflammatory activity. The objective of this study was to identify such PHGDH activators. Methods: We screened a drug repurposing library for PHGDH activators and assessed their antiseizure and anti-inflammatory properties using various zebrafish and mouse epilepsy models and explored the mechanistic consequences of activating PHGDH in a cell line, in astrocytes, and in zebrafish heads. Finally, we assessed the efficacy of clioquinol as add-on treatment in three severe DRE patients in a clinical open pilot proof-of-concept study. Results: We identified haloquinolines from a drug repurposing library as potent activators of PHGDH. The most promising haloquinoline clioquinol can increase the catalytic activity of PHGDH up to 2.5-fold, thereby increasing de novo glycine biosynthesis and resulting in reduced glutamate levels. Moreover, we show that clioquinol has PHGDH-dependent antiseizure activity as well as anti-inflammatory properties in vivo using various zebrafish and mouse epilepsy models. Finally, we demonstrate the efficacy of clioquinol as add-on treatment in severe DRE patients; two patients showed a 37%-47% reduction in seizure frequency, and all three patients noted a positive impact on quality of life and seizure severity.
Commentary
A treatment for drug-resistant epilepsy (DRE) is to switch glucose metabolism from glycolysis to a ketogenic pathway.1,2 Thevissen et al 3 suggest something different, increasing glycolysis. The authors suggest increasing a step in glycolysis that leads to serine and glycine synthesis, the conversion of 3 phosphoglycerate to 3-phosphonohydroxypyruvate (also known as 3-phosphooxypyruvate; Figure 1). The enzyme that is responsible, phosphoglycerate dehydrogenase (PHDGH) is very important to activate. Enhancing PHDGH increases the next step of pathway, which uses glutamate as a nitrogen donor. The result is a decrease in glutamate levels and increased serine and glycine (Figure 1). Targeting PHDGH is also logical because low PHDGH and PHDGH mutations are associated with epilepsy. In addition, PHDGH is primarily expressed in astrocytes and increasing PHDGH leads to an altered state of astrocytes that is anti-inflammatory, which ameliorates epilepsy. 4
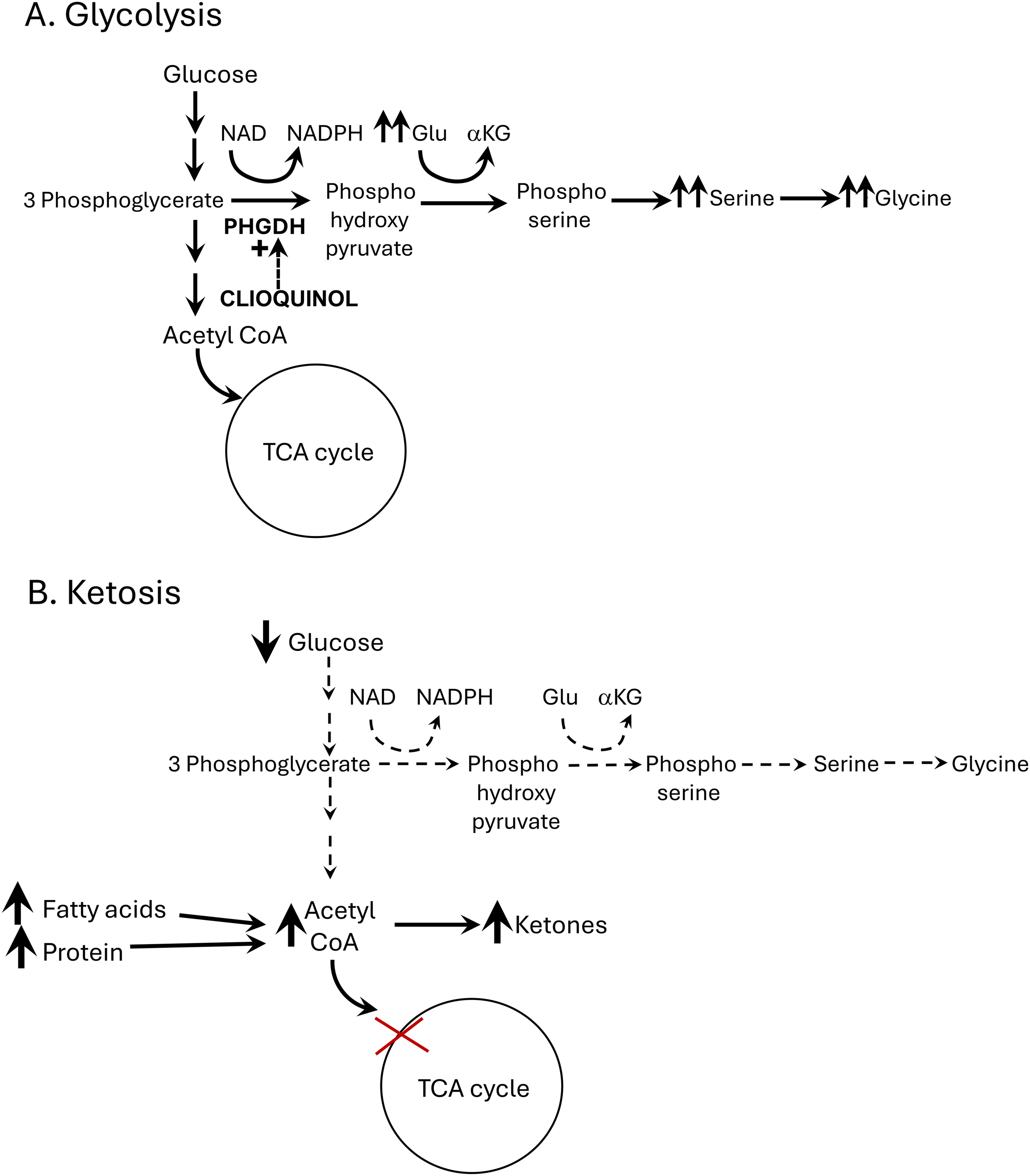
When glycolysis helps and when it exacerbates epilepsy. (A) When glycolysis improves epilepsy. Glycolysis can be valuable because the serine/glycine pathway is promoted. A step in the pathway produces 3 phosphoglycerate which leads to serine/glycine synthesis. By activating the enzyme phosphoglycerate dehydrogenase (PHDGH), glutamate levels may be reduced and both serine and glycine should be increased (double arrows). Thevissen et al 3 provide evidence that clioquinol (dotted arrow) can activate PHDGH and therefore may be an antiseizure therapy in epilepsy, possibly by reducing glutamate, increasing glycine, or other mechanisms. (B) When glycolysis exacerbates epilepsy. Individuals who have DRE often benefit from the ketogenic diet, which reduces carbohydrates and therefore reduces glycolysis. One outcome is the tricarboxylic acid (TCA) cycle is reduced as oxaloacetate is shunted to gluconeogenesis, leading to increased acetyl CoA. Another outcome is dietary fats and proteins, which replace carbohydrates, are metabolized to acetyl CoA. Both outcomes lead to an accumulation of acetyl CoA which drives formation of ketones.
The authors identified haloquinones as potential agents that increase PHDGH activity. Clioquinol was one, although historically it has been used only as an antifungal and antibiotic medication. To test clioquinol and other haloquinones they used several approaches, a strength of their study: cell lines, zebrafish, mice and patients with DRE.
They first showed that clioquinol increased PHDGH activity, and that clioquinol failed to do so in the presence of a PHDGH inhibitor. For these experiments they assayed NADPH as a reflection of PHDGH activity (Figure 1). Then they used a breast cancer cell line and clioquinol was added after depletion of serine and glycine. Metabolites were measured by gas chromatography-mass spectrometry and the results showed decreased glutamate and increased glycine after exposure to clioquinol. These data supported their hypothesis, although they also expected increased serine. Next they used human induced pluripotent stem cell-derived astrocytes and found that clioquinol increased glycine, although there was only a trend for a decrease in glutamate and no significant effects on serine. Although it is important to address astrocytes, it would have been valuable to include reactive astrocytes because they are important to epilepsy and implicated in DRE.
In zebrafish larvae, they first showed that exposure to a PHDGH inhibitor led to epileptiform events detected using local field potential recordings. Then they used two models of hyperexcitability to examine effects of clioquinol. The first used ethyl ketopentanoate, an inhibitor of glutamic acid decarboxylase that induces epileptiform activity. They also used the scn1lab−/− strain, which has spontaneous seizures and simulates Dravet syndrome. Locomotor activity and several measurements of seizures were made, a strength of the analyses. They also assayed glutamate from zebrafish heads. Together, the results clearly showed clioquinol reduced seizure-like phenomena and glutamate, although there was no effect on serine or glycine.
In the 6-Hz test, seizure duration was reduced by clioquinol, suggesting an antiseizure effect not only in zebrafish but also in mice. Then they elicited self-sustained SE and examined inflammatory markers in hippocampus 1 week later. Injections of vehicle or clioquinol (2x/day starting the day after SE) were made until the first week ended. Interestingly they selected a low dose of clioquinol, which had not shown an effect in the 6-Hz test. Also interesting, they found increased PHDGH gene expression. They were expecting increased PHDGH activity, but not necessarily gene expression. The main finding was reduced inflammatory markers, although not all inflammatory markers were reduced. At this point their hypotheses were generally supported. One might expect that they would use the self-sustaining SE model to conduct a study of chronic seizures but instead they took the opportunity to treat three adolescents with DRE, defining DRE as failing 3 antiseizure medications. All had Lennox–Gastaut syndrome. The individuals were treated with a low dose for 2 weeks and then a higher dose for 6 weeks. Two patients showed reduced seizure frequency and additional signs of improvement, such as reduced severity of seizures using a National Hospital Seizure Severity scale, and increased quality of life based on a Personal Impact of Epilepsy scale. The other patient did not show a detectable benefit of treatment. However, blood levels of clioquinol appeared to be low in all individuals, suggesting a very low dose had been achieved and a greater effect might occur with a higher dose. Although preliminary, the results are indeed suggestive that clioquinol is valuable to study further, which is what the authors conclude. To their credit, they state the work is suggestive and more needs to be done.
The patients showed no signs of toxicity, which is important because clioquinol is known to exhibit toxicity in animals and humans, which the authors mention. After its use as an antibiotic, use of clioquinol stopped because some individuals developed subacute myelooptic neuropathy (SMON), leading to vision loss and other disturbances. 5 For many years it was not clear why SMON occurred, but it was mainly in Japan, suggesting a genetic cause. A recent study suggests a common polymorphism in the Japanese population may be the reason. The study used zebrafish to first show clioquinol led to vision loss and systemic toxicity. They then used an innate antioxidant highly expressed in the eye, NQO1, to block the toxicity. Evidently in Japan there is a common polymorphism that inactivates this innate protection. 6 This is just one study of many in a very large literature about clioquinol toxicity.5,7
An intriguing aspect of the work by Thevissen et al 3 is how clioquinol exerted its actions. Although reducing glutamate levels is often suggested as a way to reduce seizures, reducing glutamate might reduce excitation of GABAergic neurons, causing more seizures rather than less. It is also interesting to consider the effects of raising serine and glycine levels. If they were increased, NMDA receptors might be more active and that would promote excitation rather than inhibit it. On the other hand, glycine can have inhibitory actions.
Clioquinol also has other effects unrelated to PHDGH. For example, clioquinol chelates zinc, and in rodent hippocampus, an injection of clioquinol depletes zinc transiently. 8 At high doses, hippocampal neuronal activity increases (assayed using the neuronal activity marker c-Fos). After c-Fos is induced, it is followed by a pattern of cell loss that resembles hippocampal sclerosis. The authors of the study8 noted that clioquinol is associated with transient global amnesia and the depletion of hippocampal zinc and hippocampal neuronal loss may provide an explanation. However, amnesia was not mentioned by Thevissen et al 3 in their patient sample, possibly because the clioquinol doses they used were low. Notably, hippocampal and amygdala injury after high-dose acute clioquinol can lead to partial epilepsy years later. 9 Thus, the study of Thevissen et al, 3 which potentially sweetens the pot of drugs for DRE, might be followed by an after taste.
Footnotes
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the NIH, New York State Office of Mental Health (grant numbers R01 NS106983, R37 NS126529, R01 NS106983).
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.