
Abstract
Developmental/epileptic encephalopathy with spike-wave activation in sleep (DEE-SWAS) is newly proposed nomenclature put forth by the International League Against Epilepsy (ILAE) to replace the problematic electrographic and/or clinical phenotypes of electrical status epilepticus of sleep (ESES) and continuous spike-wave in sleep (CSWS). The nomenclature update represents a noble effort to minimize the confusion of how to define and appropriately utilize this alphabet soup of acronyms, thereby aiding in future clinical and research efforts. The name change fails to capture greater challenges within the field, which still plague diagnosis and treatment and stagnates substantive research advancements. Through a directed literature review of DEE-SWAS with emphasis on the new ILAE nomenclature and the RESCUE-ESES trial, we will highlight major persistent quandaries in the field. These include inadequate or insufficient diagnostic biomarkers (ie, the spike wave index), the highly variable clinical manifestations, ranging from dubious associations to profound developmental regression, presumptively caused by spike-wave activation in sleep, and variable and often ineffective treatment paradigms. We will also review the broader diagnostic evaluation of DEE-SWAS. By doing so, we aim to shed light on crucial research and clinical questions that could advance our understanding of diagnosing and treating children with DEE-SWAS, as well as addressing the uncertainty surrounding the neurological effects of sleep-activated epileptiform discharges.
Keywords
Introduction
In 1971, Patry et al introduced the concept of electrical status epilepticus of sleep (ESES) in six children with severe neurodevelopmental delays and robust activation of epileptiform discharges in slow wave sleep. 1 A spike-wave index (SWI) (the number of 1-s bins containing a spike-wave discharge divided by overall time assessed) of ≥85% was observed in each child. For many, ESES describes an EEG, whereas continuous spike-wave in sleep (CSWS) and Landau-Kleffner Syndrome (LKS) describe distinct clinical phenotypes, but ESES and CSWS have been used interchangeably to describe clinical and electrographic phenotypes.
Challenges in terminology persisted despite efforts to align definitions.2–5 In 2013, a survey of 219 neurologists and epileptologists showed that 59% thought ESES and CSWS were synonymous. Most considered a SWI ≥85% to be a diagnostic cutoff, however there was notable variation in how to calculate the SWI. The only clear region of agreement (87%) was its presentation in children between 3 and 12 years. 3
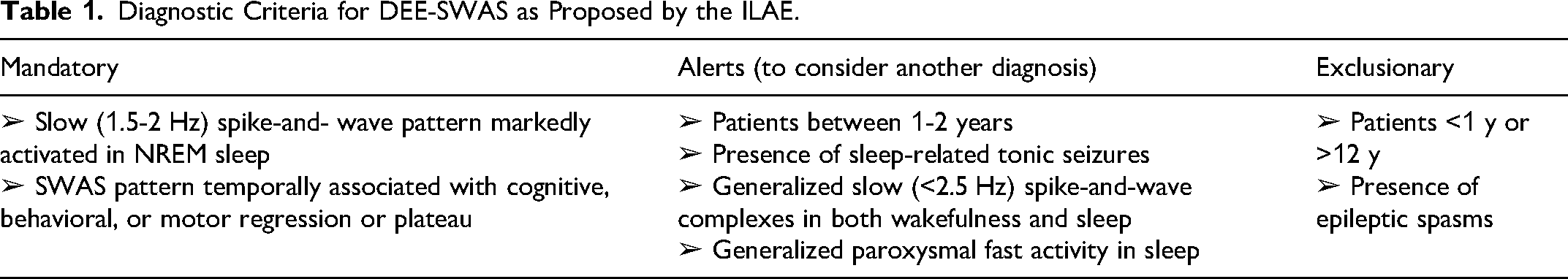
In 2022, the International League Against Epilepsy (ILAE) proposed new terminology for a host of epilepsy syndromes. 6 Any ESES-related disorder falls under Developmental/Epileptic Encephalopathy with Spike-Wave Activation in Sleep (DEE-SWAS). The epileptic agnosia phenotype of LKS remains a clinically distinct subtype. Diagnostic criteria for DEE-SWAS as proposed by the ILAE are shown in Table 1. DEE-SWAS is defined by a pre-existing neurodevelopmental disorder whereas EE-SWAS has preceding (near-)normal development; otherwise EEG features, diagnostics, and treatment considerations do not significantly differ.
Diagnostic Criteria for DEE-SWAS as Proposed by the ILAE.
Many unanswered clinical questions remain. We will highlight key clinical and research quandaries DEE-SWAS poses, including: 1) what neurophysiological biomarkers best define DEE-SWAS?; 2) how can we reliably define and measure regression or plateau?; 3) what additional testing aids in the DEE-SWAS diagnosis?; and 4) what are the best available treatments for DEE-SWAS?
The Role of EEG in Diagnosis of DEE-SWAS
The primary biomarker for DEE-SWAS is the SWI. The historical 85% cutoff has been predominant, but proposed guidelines in 2009 offered a lower cutoff of ≥50% in NREM sleep. 7 Some groups utilize a 25% cutoff. 8 The ILAE did not specify a SWI cutoff, stating “lower percentages may also be associated with significant regression in cognitive or behavioral function.”
There is no evidence to support a specific SWI to qualify as SWAS. There is no established percentage increase in SWI between wakefulness and sleep, nor definition that encapsulates changes in morphology, distribution, or frequency. The following questions highlight this ambiguity:
Does a SWI of 40% in wakefulness increasing to 55% in sleep count as SWAS? Does a SWI of 80% in wakefulness increasing to 100% in sleep count as SWAS? Is it SWAS if abundant focal discharges in wakefulness become bilateral and synchronous in sleep?
There is a lack of agreement about the topographical distribution of SWAS. This leads to important clinical questions:
Should spike-waves only be counted if they become bilateral and synchronous in sleep (Figure 1) or can they be focal (Figure 2) or multifocal? If there are multifocal spike-waves, does each focus count as an independent SWI OR are the number of bins with a spike, regardless of location, counted (Figure 3)?
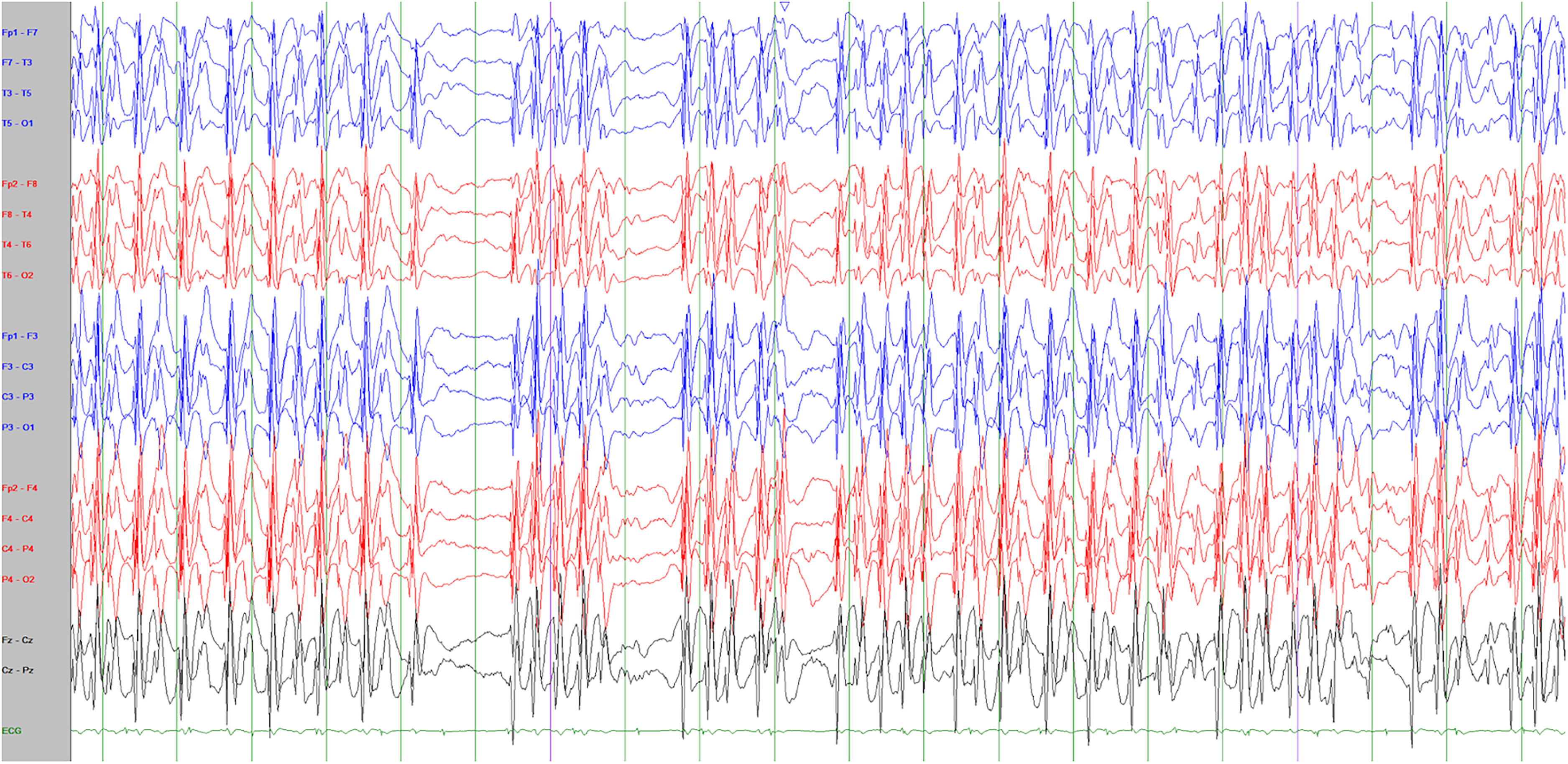
Anterior-posterior bipolar montage of a 20-s EEG epoch showing robust bilaterally synchronous, generalized, spike-wave discharges at a 1.5-2 Hz frequency. While there are brief segments of relative voltage attenuation and spike quiescence, the spike-wave index is 100%.
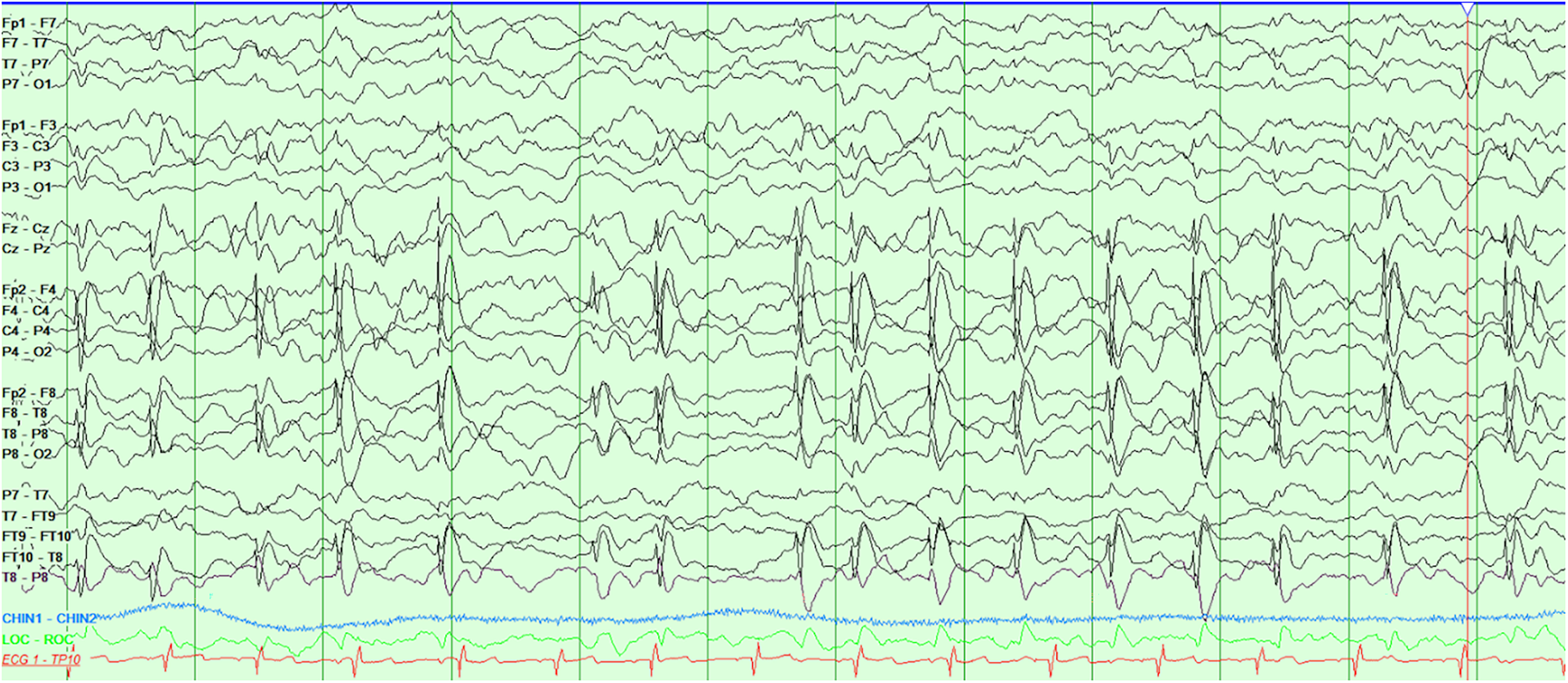
Anterior-posterior bipolar montage of a 12-s EEG epoch demonstrating high-voltage 1.5-2 Hz spike-wave discharges from the right centrotemporal (C4/T8 maximal) region. The spike-wave index for this epoch is 11/12, or 91.7%.
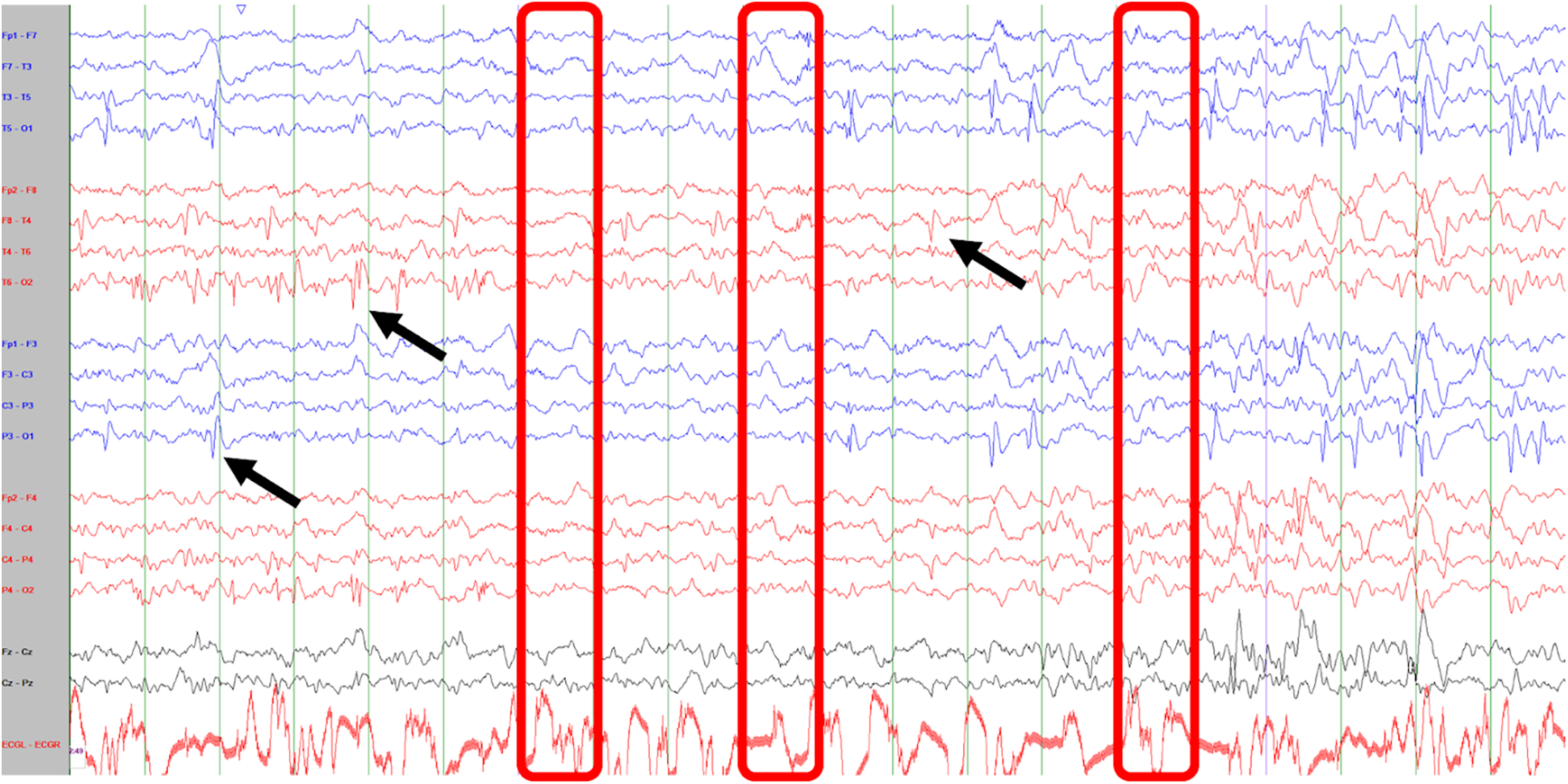
Anterior-posterior bipolar montage of a 20-s eeg epoch of sleep with three independent spike populations (arrows define populations at left posterior quadrant [P3-O1/T5-O1], right posterior quadrant [P4-O2/T6-O2], and right centrotemporal region [C4-T4]). Using a standard spike-wave index calculation, only three 1-s bins (boxes) do not have clearly identifiable epileptiform discharges, accounting for a potential index of 17/20 = 85%. Another perspective is to calculate the SWI for each spike population: P3-T5-O1 = 45% (9/20); P4-T6-O2 = 35% (7/20); C4-T4 = 55% (11/20).
Beyond the above questions, the following irksome problems plague EEG readers:
Problem: Quantifying the SWI over an entire night is time-consuming. Response: methodologies counting the first 100 or 300 s of NREM sleep after the first sleep spindle have been reported.9–11 Automated software has shown non-inferiority to human scorers.12,13 However, a survey of 69 epileptologists revealed substantial variability in methodology: 37.6% assess the first five minutes of sleep, 27.5% assess the first 100 s of sleep, 15.9% have no standardized method, and 18.8% use mixed approaches. 14
Problem: The relevance of sleep stage in SWI calculation remains unclear. Response: Different patterns of SWAS have been described, including a slow wave sleep pattern, a light sleep pattern, and a continuous pattern. 15 These authors argued “shortcut” sampling methods (<30 min) may be inaccurate. It remains unknown whether EEG pattern correlates with clinical phenotype, severity, or treatment response.
Problem: The ILAE suggested EEG criteria for DEE-SWAS lack evidence and may cause confusion. Response: The ILAE Task Force proposed a mandatory 1.5-2 Hz slow spike-and-wave pattern as a diagnostic feature, which is “usually diffuse but may occur more focally (typically frontally) or multifocally.” 6 This is the first inclusion of a specific frequency to the EEG offered in the literature. This may lead to confusion with the slow spike-and-wave pattern of Lennox-Gastaut syndrome (LGS), which the ILAE flags for differential consideration (Table 1).
Problem: The SWI may not be the ideal EEG biomarker. Response: For better or worse, SWI remains the best studied and easily measured biomarker. Some researchers have advocated for assessing spike-wave frequency or density, ie, the number of spikes per unit of time. 16 This measure has not been well utilized. The sleep spindle is an attractive biomarker for DEE-SWAS warranting further study. Figure 4 highlights key considerations on how spindles may relate to DEE-SWAS.
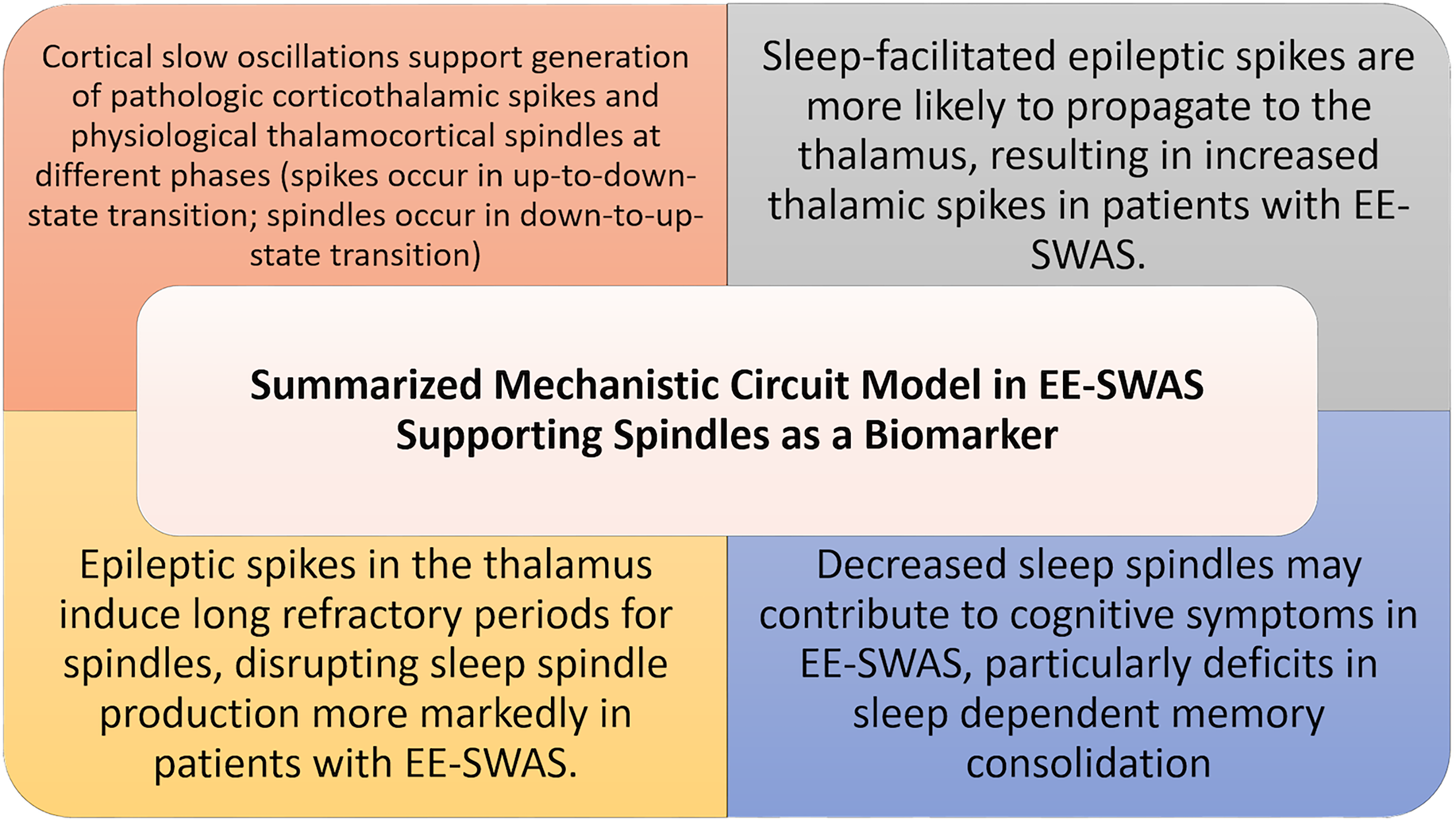
Summarized mechanistic circuit model in EE-SWAS supporting spindles as a potential biomarker. 17
What Is the Clinical Significance of SWAS?
Language is the most commonly impacted domain in DEE-SWAS. Language regression occurs with onset of SWAS, but premorbid language delays are possible. 18 Regression can fluctuate and appears to correlate with SWAS and treatment response. The ILAE defines LKS as a distinct EE-SWAS subtype involving loss of receptive language skills preceding expressive language declines in children with (near-)normal development.6,19
There is considerable clinical variability in DEE-SWAS. Global neuropsychological impairment is not uncommon.18,20 Cognitive functions impacted may include general intellectual functioning, attention, executive functioning, cognitive efficiency, visuospatial skills, learning/memory, and academic achievement.8,20–23 Behavioral regression is observed in approximately 50%, including mood changes, aggression, autistic features, and changes to eating and sleeping.18,24,25 Motor deterioration may manifest as ataxia, dyspraxia, dystonia, hyperkinesia, and unilateral deficits.26–29 While seizures and SWAS typically remit in adolescence, unfavorable outcomes with persistent deficits are associated with younger age of diagnosis, longer disease duration, lower IQ at time of diagnosis, refractoriness to interventions, and etiology.24,28,30
A systematic review of DEE-SWAS found that 63% of cases were consistent with DEE-SWAS, 20% of cases were consistent with EE-SWAS, and 17% of cases were not able to be classified. 31 Both groups showed similar patterns of regression at onset of SWAS. The DEE-SWAS group was more likely to present with a genetic etiology, earlier age of seizure onset, and lower functioning prior to further regression.
The ILAE nomenclature task force highlights that neurodevelopmental consequences are expected within weeks from SWAS onset. This does not address a scenario of incidental discovery of SWAS in a child with longstanding neurodevelopmental delays or autism without historical or recent regression or plateau. This difficult clinical area desperately warrants further study.
Neurodiagnostic Considerations in DEE-SWAS Beyond the EEG
Many developmental brain abnormalities have been associated with DEE-SWAS, in particular perinatal thalamic injuries and perisylvian polymicrogyria. Patients with prominent sleep-potentiated epileptiform activity were 7.5x more likely to have early developmental or thalamic lesions compared to controls. 32 Neonatal thalamic hemorrhage has been particularly associated with DEE-SWAS. 33
Reports link multiple genes to DEE-SWAS. There is no unifying genetic etiology, but certain monogenic and chromosomal changes appear more common in DEE-SWAS. Channelopathies are the most common underlying pathway. Genetic testing may support “precision medicine” efforts such as utilizing memantine in patients with GRIN2A pathogenic variants, and primidone in patients with TRPM3 pathogenic variants.34–36
Figure 5 provides a summary of neurodiagnostic considerations in DEE-SWAS. DEE-SWAS likely arises much like infantile epileptic spasms syndrome (IESS) or LGS, through multitudinous pathways creating a perturbed epileptogenic and encephalopathic network.

Diagnostic evaluations after a diagnosis of DEE-SWAS is made. Many genetic abnormalities may contribute to the DEE-SWAS phenotype and whole exome or whole genome sequencing (WES or WGS) are increasingly recommended. Chromosomal microarray retains utility looking for large deletions and copy number variants. A thoughtful clinical history may uncover a possible TORCH infection as an underlying etiology or iatrogenic source such as an offending antiseizure medication. Less commonly assessed but an area of active research is assessment of Interleukin levels.37,38 CBZ: carbamazepine; OXC: oxcarbazezpine; CMV: cytomegalovirus.
Treatment of the EEG to Treat the Patient
Reports describe successful treatment with multiple therapies. Figure 6 highlights some of the most commonly explored therapeutic options. A pooled analysis of 575 cases of treated ESES found improvements were most commonly observed with surgery (90%), steroids (81%), or benzodiazepines (68%); standard antiseizure medications were least effective (49%). 39
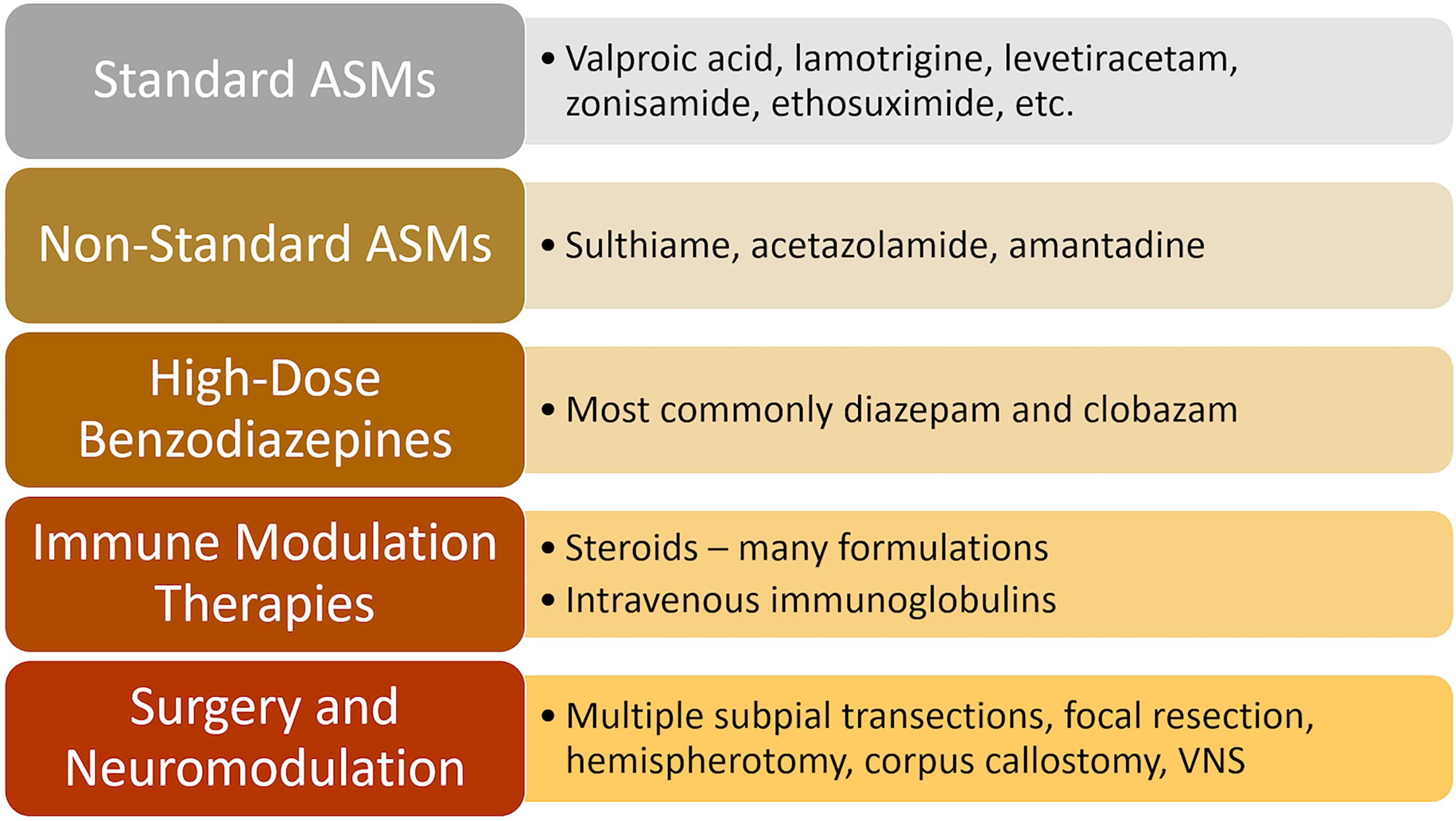
Common treatment considerations in DEE-SWAS.
Benzodiazepines Versus Steroids
No data define the most effective benzodiazepine or steroid regimen. The most commonly utilized benzodiazepines include diazepam and clobazam.16,40 There is no direct comparison of these treatments and little to guide duration. Surveys in 2014 and 2021 suggested that benzodiazepines were the most-common first choice, with diazepam being more commonly chosen than clobazam.41,42
Reported steroid regimens include dexamethasone, hydrocortisone, adrenocorticotropic hormone, prednisone, and intravenous methylprednisolone.43–45 The most efficacious formulation, method of delivery (eg, oral daily vs intravenous pulse), or duration of therapy have not been established.
The RESCUE-ESES trial was unable to provide a definitive answer to the steroid versus benzodiazepine debate due to early termination of the study secondary to poor enrollment. This study enrolled only 45 participants across 8 European sites over 7 years. 46 The trial compared clobazam to daily oral prednisolone or pulse intravenous methylprednisolone over 6 months with primary endpoints of intelligence quotient (IQ) and cognitive sum score responder rates. There was an improvement in IQ in 25% of children receiving steroids, not observed with clobazam, however this change was not reflected in the cognitive sum score nor associated with EEG improvement. Change in SWI, EEG responder rate (a decrease in SWI ≥25% from baseline), and patients with SWI <50% were not significantly different between groups at 6 and 18 month timepoints. Frequency of side effects was comparable between groups, including 45% of children receiving steroids and 52% of children receiving clobazam. The authors argue, even in the under-powered nature of the study, that the IQ change is enough to support early corticosteroid treatment over clobazam. 46
Epilepsy Surgery and Neuromodulation Therapies
When interictal activity is causing an epileptic encephalopathy, surgery should be explored, as it may improve cognition. 47 Surgical workup is generally not considered in DEE-SWAS in the absence of concurrent refractory focal epilepsy, however, focal epilepsy surgery, can resolve ESES and improve cognition in patients with unilateral structural etiology. Given malformations of cortical development and thalamic injury are the most common structural etiologies, if a disconnection surgery, such as hemispherotomy, is determined medically necessary in appropriately selected patients, an approach with thalamic disconnection may be necessary for successful treatment. 48
Neuromodulation for DEE-SWAS has yet to be formally investigated, with forthcoming clinical trials anticipated. Evidence is limited to case reports, such as with VNS. 49 Patients with refractory DEE-SWAS without clear resective or ablative targets make thalamic neurostimulation an attractive consideration, although further studies are needed to determine their safety and efficacy.
Defining Treatment Success
There are nuances in defining treatment success. SWI “percent reduction” (ie, a 50% reduction in a SWI of 80% would reduce to 40%) and SWI “absolute reduction” (ie, a 50% reduction in a SWI of 80% would reduce to 30%) differ. It's unknown if one is more meaningful. The RESCUE-ESES trial defined an “EEG responder” as an absolute reduction of ≥25%. 46 Another nuance to consider is when bilateral synchronous SWAS becomes focal after treatment. Perhaps the best definition of success is clinical improvement associated directly with electrographic improvement.
Due to the many limitations in neuropsychological testing, clinical improvement is subjective. Challenges persist in cases where the SWI improves but the patient does not—was the SWAS clinically significant or was the duration sufficient to cause persistent sequelae? An unexpected contrapositive is clinical improvements without appreciable change in SWI or neuropsychological testing. This may support other biomarkers (eg, sleep spindles) or parental reports as more sensitive to meaningful change, or suggests that objective neurodevelopmental assessments may be biased against identifying measurable change in this population.
Conclusions and Future Directions
The DEE-SWAS nomenclature represents an important unifying step in connecting clinicians and researchers. Our review highlights many questions that require significant equipoise to counter the paucity of evidence. Working towards unified and accepted definitions of SWAS are essential. Neurophysiological assessments beyond the SWI, including spindle metrics, require further validation.
Efforts to create a reliable inter-rater scoring system, similar to the BASED score for IESS, may assist in this. 50 Hypsarrhythmia is a variably defined electrographic phenotype with poor inter-rater reliability, echoing the DEE-SWAS SWI dilemma. The parallels between DEE-SWAS and IESS are manifold and the efforts of the IESS community to improve diagnosis and treatment paradigms should be heeded.
Azeem et al found that patients with perinatal stroke showed higher spike frequency and lower delta frequency preceding the onset of ESES. 51 Monitoring and assessing at-risk patients with serial EEGs may help predict those at risk of conversion to DEE-SWAS and initiate therapy prior to developmental regression. This could lead to efforts like the EPISTOP and PREVENT trials.52,53
The tribulations of the RESCUE ESES trial should not dissuade future involvement from clinicians and/or industry. The above efforts to improve and standardize our treatments and measurements will be significantly augmented by sufficiently powered, randomized, double-blind, controlled trials.
Footnotes
Authors’ Note
This work was completed as part of the efforts of the Pediatric Epilepsy Research Consortium (PERC) DEE-SWAS special interest group.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.