
Abstract
Mapping neuronal circuits that generate focal to bilateral tonic–clonic seizures is essential for understanding general principles of seizure propagation and modifying the risk of death and injury due to bilateral motor seizures. We used novel techniques developed over the past decade to study these circuits. We propose the general hypothesis that at the mesoscale, seizures follow anatomical projections of the seizure focus, preferentially activating more excitable neurons.
Keywords
Historical Perspective
Repeated generalized tonic–clonic seizures (GTCSs) are the single most significant risk factor for sudden unexpected death in epilepsy, SUDEP. 1 Mapping neuronal circuits that underlie GTCSs is crucial for understanding general principles of seizure propagation. Historically, John Hughlings Jackson, one of the founders of neurology and human neuroscience, first pointed out that the motor cortex generates GTCSs. 2 He thought that convulsions that appear on one side of the body result from the corresponding ictal discharge that begins in the contralateral motor region that represents that body part. Later in human studies, Wilder Penfield confirmed this using electrical stimulation in conscious epilepsy patients during surgery, mapping the motor cortex and delineating the homunculus. 3 Herbert Jasper and Penfield collaborated to localize seizure foci to the area of electroencephalogram (EEG) abnormalities. 4 They studied cortical function and seizure networks by electrically stimulating various brain regions. They showed seizure-induced reorganization of the motor homunculus in epilepsy patients. 5 In 1954, they brought together the conclusions of their studies in the landmark book, Epilepsy and the Functional Anatomy of the Human Brain, 5 helping us localize the site of seizure onset by evaluating the seizure semiology and intracranial and scalp EEG findings.
The techniques continued to improve, allowing visualization of activated brain regions. In the 1970s, Louis Sokoloff developed 2-deoxy-D-[ 14 C]glucose (2-DG) autoradiography to monitor metabolic acitivity. 6 The radioactive analog of glucose stops at the second step of glycolysis, accumulating in the metabolically active somas, creating a functional activity map of activated neurons. The 2-DG mapping technique was instrumental in studying the functional anatomy of temporal lobe seizures, leading to the dentate gate hypothesis that states propagation of seizures in the hippocampus is gated by strong inhibition of the dentate gyrus. 7 This hypothesis led to explosive growth in the field, generating more than 2500 scientific papers on dentate gyrus and epilepsy. Other techniques, such as single-photon emission computed tomography, visualize cerebral blood flow changes during seizures after delivery of a gamma-emitting radioisotope, revealing the most affected brain regions in humans. 8 The advent of c-Fos immunohistochemistry provided cellular resolution. Yet, whole-brain seizure circuit mapping at single-cell resolution remained challenging due to the lack of computerized image acquisition, processing, and analysis.
Many recent technological advances, including those driven by the Brain Research Through Advancing Innovative Neurotechnologies Initiative of NIH, developed novel tools and techniques like activity reporter mice, tissue clearing, and automated microscopy, including super-resolution and light-sheet microscopy, that generate high-resolution, high-throughput imaging. These techniques allow 3D reconstruction of images to visualize activated neuronal circuits. Conditional knockout mice and Cre-driven viral tracing target specific neurons, and chemo- and closed-loop optogenetic techniques suppress specific structures precisely, elucidating their function.
Temporal and frontal lobe seizures are the 2 most common focal onset types of seizures that lead to GTCSs. Temporal lobe seizures have been mapped extensively. However, the functional anatomy of frontal lobe seizures was studied less well. Using the techniques described above, we investigated the circuits that mediate focal onset seizures and general principles that underlie seizure propagation.
Frontal Lobe Onset Seizures
The frontal lobe, specifically the motor cortex, sends extensive projections to the basal ganglia, which consists of the striatum, globus pallidus, subthalamic nucleus, and the substantia nigra pars reticulata (SNR). 9 Many previous studies have demonstrated electrographic activation of the basal ganglia structures during seizures. 10 -16 Drug experiments such as GABAergic inhibition of the SNR 14 or pharmacological activation of the striatum 17 exhibited anticonvulsant effects, demonstrating that the basal ganglia modulate seizures. Yet, direct cellular evidence of basal ganglia circuit activation was lacking. We created a seizure focus in the premotor cortex by placing cobalt there. This model provided reliable, spontaneous seizures without introducing a bias toward a specific cortical layer in which seizures originate. We also used the activity reporter TRAP2 mice that express a fluorescent protein under the control of the c-Fos promoter to label seizure-activated neurons.
Frontal lobe-onset focal to bilateral seizures propagate through the basal ganglia and motor thalamic nuclei on the mesoscale (structural) and cellular levels. 18 We hypothesized that anatomical projections of the seizure focus shape the seizure circuit, so we tracked efferent projections of the seizure focus in the premotor cortex with the AAV9 GFP virus and labeled seizure-activated axons in TRAP2 mice simultaneously. Using microscopy and 3D reconstruction, we found that seizures propagate subcortically through the basal ganglia, using anatomical projections of the seizure focus.
The striatum consists of indirect and direct pathway neurons with different projections. 19 The indirect pathway medium spiny neurons express dopamine D2 receptors (D2 R) on their surface and project sequentially through the globus pallidus externus, subthalamic nucleus, and SNR, inhibiting thalamic nuclei and voluntary movement. The direct pathway medium spiny neurons express D1 receptors (D1 R) and project to the globus pallidus internus and SNR, disinhibiting the thalamic targets and facilitating voluntary movement. 19 Previous studies did not indicate whether seizures preferentially activate the indirect or direct basal ganglia pathway. We found seizures preferentially activated the indirect pathway 18 because 80% of all activated neurons in the striatum were D2R-expressing. Infusion of sumanirole, a D2 R agonist, directly into the striatum and its systemic administration transiently abolished seizure activity, similar to Turski et al findings that showed striatal D2 R agonist infusion also protected rats against pilocarpine limbic seizures. 20 D2R-expressing indirect pathway neurons are more excitable than the direct pathway neurons, with a smaller surface area, smaller dendritic trees, and a less hyperpolarized resting membrane potential, 21,22 explaining their preferential activation. These studies suggest that in addition to the connectivity of the seizure focus, neuronal excitability also shapes the seizure circuit.
This study mapped neuronal activation of the basal ganglia during frontal lobe onset seizures on the cellular level. In particular, we show preferential activation of the indirect pathway neurons in the striatum, which are more excitable than the direct pathway neurons. Seizures follow anatomical connections of the seizure focus, activating more excitable neurons. Therefore, multiple subcortical structures of the basal ganglia could be targeted for modulation and stimulation. For example, deep brain stimulation of the subthalamic nucleus, a part of the indirect pathway, decreases the activity of SNR and reduces seizure frequency in patients with drug-resistant epilepsy. Similarly, low-frequency striatal stimulation was also anticonvulsant in 57 patients with refractory temporal lobe epilepsy. 23
Secondary Bilateral Synchrony During Seizures
We further investigated the mechanisms of bilateral synchronous cortical activity during frontal lobe focal to bilateral tonic–clonic seizures, previously known as secondarily generalized. 24 The canonical centrencephalic model, first proposed by Penfield, places the diencephalon at the center of bilateral synchronization. 5,25 -29 During natural sleep, the thalamus generates bilateral spindle oscillations of 7 to 14 Hz, which are thought to underlie the spike-and-wave discharges observed in primarily generalized absence seizures. 28,30 It is proposed that focal seizures reach the centrencephalic system leading to generalized seizures. However, classical callosotomy studies suggest the corpus callosum mediates bilateral synchrony and seizure spread. 31 To disentangle the roles of the thalamus and callosum in bilateral seizure spread, we recorded local field potentials (LFPs) from the bilateral premotor cortex and ventrolateral motor thalamus simultaneously to map their temporal sequence of activation. 24 We selected the motor thalamus because it receives strong projections from the right motor cortex, the seizure focus in our case. 32 Seizures that originated in the right premotor cortex reached the left premotor cortex faster than they did the left thalamus.
We also suppressed the right motor thalamic nuclei using chemo- and closed-loop optogenetics and lesioned the corpus callosum. 24 Suppression of the motor thalamus did not prevent bilateral spread, whereas callosal lesioning abolished secondary generalization. Using viral tracing, we found that the right and left thalami have minimal direct monosynaptic commissural connections, whereas the callosum sends extensive contralateral projections that are transmitting seizures. Thus, the circuits of focal to bilateral seizures are very different from those of primarily generalized absence seizures, which affect the somatosensory cortex and its thalamocortical projections to the sensory thalamic nuclei. 33 We could not confirm the existence of a single seizure choke point and found that cortical integration of seizure activity is independent of thalamic input. Thalamocortical oscillations amplified focal onset seizures, while the corpus callosum spread them bilaterally.
The Motor Cortex Is Active During GTCS
The motor cortex was considered the key structure that generates GTCSs, 2 yet recent studies raised doubts about its activation during GTCSs. It has been proposed that GTCSs arise from focal seizures accessing centrencephalic structures that consist of the upper brainstem and thalamus (including the thalamocortical projections to the somatosensory cortex). 25,27 In several previous studies, motor cortical fields in the prefrontal and frontal cortex did not display increased radiolabeled 2-DG uptake or c-Fos immunoreactivity during the prolonged seizures observed in status epilepticus (SE). 34 In other studies, variable activation of the motor cortex was reported. 35,36 Studies imaging cerebral blood flow in patients with GTCSs have yielded mixed results, showing increased and decreased activation of the motor cortex. 8 However, in addition to the pioneering work of Jackson, Penfield, and Jasper, some studies in animal models showed that kindled and recurrent spontaneous seizures reorganize the functional map of the motor cortex. 37 -39
We found cortical motor activation during frontal lobe GTCSs on the cellular level. The motor cortex sends extensive projections to the basal ganglia. 9 Using viral tracing, activity reporter mice, and super-resolution microscopy, we found that frontal lobe-onset GTCSs follow anatomical projections of the motor cortex to the basal ganglia 18 and across the corpus callosum contralaterally. 24 The hemisphere ipsilateral to the seizure focus had more activated neurons than the contralateral, and superficial cortical layers 2/3 were more activated than deep layers 5/6 (Figure 1).
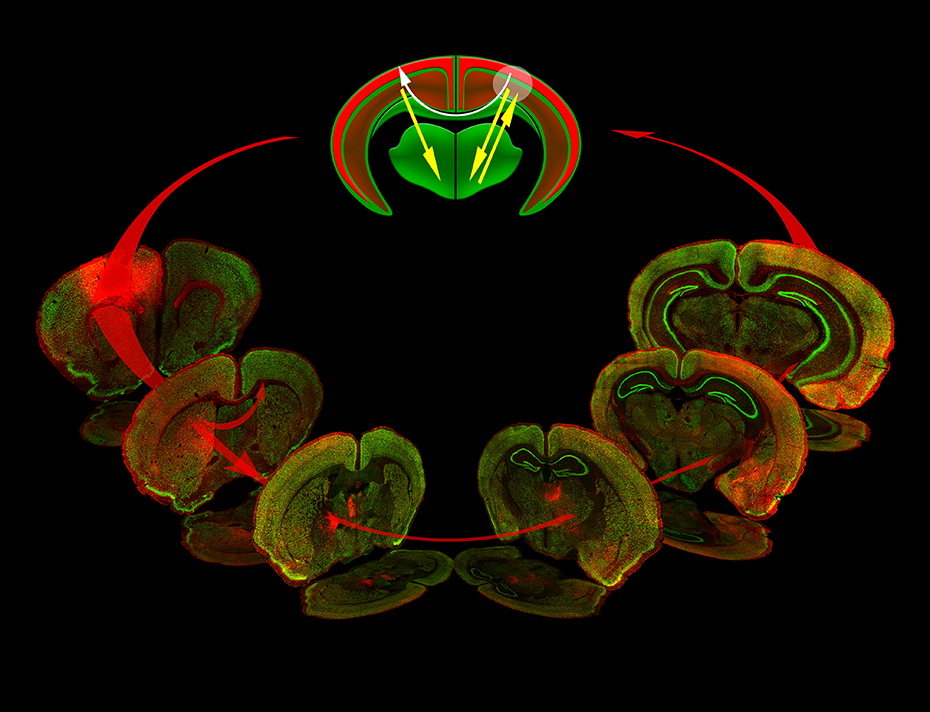
Circuit map of frontal lobe-onset focal to bilateral seizures. Seizures that originate in the frontal lobe motor cortex (a white circle) propagate subcortically through the indirect basal ganglia pathway (confocal images, red arrows): the striatum, globus pallidus externus, subthalamic nucleus, substantia nigra reticulata, and the motor thalamic nuclei. The bilateral spread occurs via the corpus callosum (top schematic, white arrow), while the thalamus amplifies seizures (top schematic, yellow arrows). The superficial cortical layers 2/3 are more activated during seizures than deep layers 5/6.
Neocortical injury-induced SE also intensely activated the motor cortex ipsilateral to the seizure focus, whereas subcortical structures such as the thalamus, hypothalamus, hippocampus, and dentate gyrus exhibited sparse labeling in the early stages and more robust staining during late-stage SE. 40 The continuous hippocampal stimulation drives hippocampal-onset generalized convulsive SE and leads to the activation of the bilateral motor cortex. 41 Neonatal hypoxic-ischemic seizures similarly exhibited symmetric bilateral motor cortical activation, whereas activation of other regions such as the somatosensory cortex, olfactory system, lateral thalamus, and hippocampal-parahippocampal structures was asymmetric. 42 In all these studies, superficial cortical layers 2/3 were also more activated bilaterally compared to deep layers 5/6, which is best explained by the anatomy and connectivity of the motor cortex.
The layered motor cortical organization allows independent, parallel transfer of neuronal activity subcortically and contralaterally. The motor cortex is an agranular cortex that lacks layer 4 and consists of 3 types of neurons: 43 intratelencephalic-type (layers 2/3, 5, 6) neurons project corticocortically and across the corpus callosum contralaterally; pyramidal tract-type neurons (layer 5) project to the brainstem; and corticothalamic-type neurons (layer 6) project to the ipsilateral thalamus. Eighty percent of all callosal axons originate from layer 2/3. 44 Therefore, stronger activation of more superficial cortical layers during hypoxia-ischemia-induced seizures in neonatal mice 42 and generalized convulsive SE 41 results from cortico-cortical axons synapsing on the superficial layers. Furthermore, the motor cortex is proposed to have top-down laminar organization, where superficial layers drive the excitation of the deep layers. 45,46 In the future, we will aim at silencing the motor cortex to stop seizures.
Excitatory Connectivity in the Hippocampus
Activation of the motor cortex, corpus callosum, and basal ganglia during motor seizures suggested that seizures spread through the anatomically connected structures to the seizure focus and between each other. We next determined whether neuronal synchrony structure depends on the direction of hippocampal excitatory anatomic connections. 47 Studies in human patients with focal epilepsy found very heterogeneous activity during a seizure. 48 Others describe that seizure propagation and synchrony are restricted to a core, with a penumbra region shaped by an inhibitory surround. 49,50 We recorded LFPs along the lamellar or septotemporal hippocampal axis during the hippocampal kindling process and used 4 measures of neuronal synchrony. Neurons linked by excitatory connections along hippocampal lamina fired synchronously during seizures. 47 In contrast, neurons along the trans-lamellar, septotemporal axis were not in synchrony during seizures (Figure 2). Kindling enhanced neuronal synchrony along the lamellar axis, while synchrony along the septotemporal axis, despite repeated stimulations, remained low. Anti-seizure drugs like phenytoin and levetiracetam diminished synchrony, demonstrating that synchrony structure depends on excitatory anatomic connectivity and plasticity.
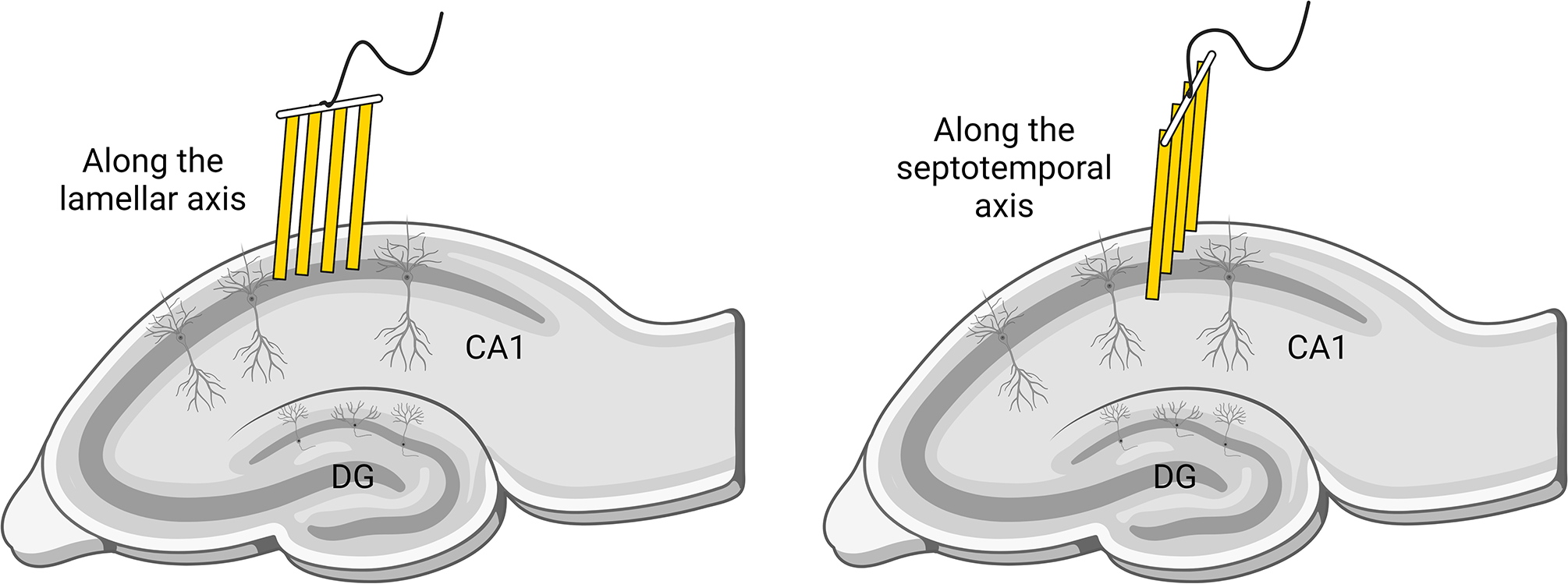
Schematic figure of the microelectrode array placement in the hippocampal CA1 cell layer.
Frontal lobe seizures activated more D2 receptor-expressing neurons than D1R-containing neurons in the striatum, even though both types of neurons receive projections from the motor cortex. 18 One explanation is that D2 receptor-containing neurons are more excitable. Therefore, we tested whether seizure-activated neurons in the hippocampus are more excitable. Previous studies testing the memory engram hypothesis suggest that more excitable cells help encode memory. Epilepsy patients often experience memory problems even without underlying hippocampal pathology. This led us to hypothesize that seizure-activated neuronal circuits overlap spatial memory engrams, leading to seizure-induced retrograde amnesia. 51 We used a T-maze spatial memory alternating task as well as activity reporter TRAP2 mice and ARC dual-labeling to visualize learning-and seizure-activated neurons simultaneously. Learning activated neurons in the CA1, 51 dentate gyrus, mediodorsal thalamus, retrosplenial cortex, and medial prefrontal cortex. 52 A seizure prevented the recall of alternation task memory and activated memory-labeled neurons and structures. Seizure-tagged CA1 pyramidal neurons were more excitable, had enlarged dendritic spines, and saturated LTP (long-term potentiation) compared to surrounding nonactive neurons. 51
Hub theory points at the existence of the superhub, a novel cell type richly connected to the rest of the network that enhances excitation and enables pathological dynamics. 53,54 We hypothesize that the pattern of neuronal activation during seizures is a stochastic process guided by the intrinsic excitability of neurons, their anatomical projections, and neuronal plasticity. It is proposed that neuronal allocation to a memory engram is a competitive process that depends on intrinsic excitability. 55 For example, overexpressing CREB (cAMP response element-binding protein) in specific neurons helps recruit them in a fear engram and increasing excitability in a random population preferentially recruits neurons into the engram. 56,57
In summary, we propose that anatomical connectivity and neuronal excitability shape seizure circuits. Further studies are needed to determine the critical nodes in these seizure circuits by opto- or chemogenetic suppression of specific structures and cell types during seizures. Furthermore, the modification of seizure circuits by the process of epileptogenesis, which involves synaptic plasticity, changes in neuronal excitability, cell loss, proliferation, and axonal reorganization, is likely to reshape seizure circuits in a dynamic fashion.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by the National Institutes of Health (R01NS120945, R37NS119012) and the UVA Brain Institute.