
Abstract
Focal cortical dysplasia (FCD) is a malformation of cortical development that is a prevalent cause of intractable epilepsy in children. Of the three FCD subtypes, understanding the etiology and pathogenesis of FCD type II has seen the most progress owing to the recent advances in identifying gene mutations along the mTOR signaling pathway as a frequent cause of this disorder. Accordingly, numerous animal models of FCD type II based on genetic manipulation of the mTOR signaling pathway have emerged to investigate the mechanisms of epileptogenesis and novel therapeutics for epilepsy. These include transgenic and in utero electroporation-based animal models. Here, we review the histopathological and electroclinical features of existing FCD type II animal models and discuss the scientific and technical considerations, clinical applications, and limitations of current models. We also highlight other models of FCD based on early life acquired factors.
Keywords
Introduction
Focal cortical dysplasia is a malformation of cortical development (MCD) caused by disrupted cortical assembly during fetal development, leading to abnormal cell migration, neuronal misplacement, and disorganized cortex with or without abnormal neuronal growth within a focal or multifocal brain region. 1 FCD is highly associated with early onset intractable epilepsy and is the most common cause of focal epilepsy requiring surgery among children. 2,3
FCD is categorized into three subtypes based on histopathological findings as defined by the International League Against Epilepsy (ILAE). 4 FCD type I is described by abnormal cortical lamination. FCD type II is defined by disrupted cortical lamination and the presence of dysmorphic neurons, either without (type IIa) or with (type IIb) the occurrence of enlarged, abnormal cells called balloon cells. The histopathological features of FCD type IIb are similar to hemimegalencephaly, a type of MCD involving a whole hemisphere, and cortical tubers in tuberous sclerosis complex (TSC). 5,6 FCD type III is characterized by cortical lamination abnormalities, as observed in FCD type I, adjacent to a principal lesion, including hippocampal sclerosis, tumor, vascular malformation, or other lesions acquired in early childhood (e.g., trauma, ischemia, encephalitis, infection).
A recent body of evidence has identified mutations in genes encoding for regulators of the mechanistic target of rapamycin complex 1 (mTORC1) signaling pathway, leading to enhanced mTORC1 signaling, as a cause for FCD type II. 7 These genes include MTOR, PIK3CA, PTEN, TSC1, TSC2, RHEB, DEPDC5, NPRL2, and NPRL3 and have been the subject of previous reviews. 7,8 The mutations are thought to occur in a subset of neural progenitor cells during embryonic development, giving rise to abnormal neurons, and in some cases, glial cells. 7,9,10 The mTORC1 pathway is a key regulator of neuron growth and development, and aberrant activation of mTORC1 signaling is thought to alter neuron structure and function, leading to pro-epileptogenic circuit formation and epilepsy. 11 The cause of FCD type I remains largely unknown, though genetic mutations have been reported in a subset of cases involving a variety of genes, including those encoding for regulators of the mTORC1 pathway, synaptic proteins, ion channels, glycosylation enzymes, and cell adhesion proteins. 12,13 The principal lesion in FCD type III likely results from acquired insults to the developing brain during pre- and perinatal stages. 4
Despite progress in understanding the etiology and pathogenesis of FCD, the cellular and molecular mechanisms leading to epilepsy is unclear and current therapeutic options for seizure control are inadequate. To address these issues, many animal models of FCD have been established to investigate the mechanisms of epileptogenesis and novel therapeutics for epilepsy. Most of these models recapitulate FCD type II by genetically targeting components of the mTORC1 pathway, either through genetic engineering or in utero electroporation (IUE) (Figure 1). There are no definite animal models of FCD type I due to insufficient knowledge about its etiology and few models of FCD type III due to its complex and ambiguous definition. In this review, we summarize the histopathological and electroclinical features of existing FCD animal models and discuss the scientific and technical considerations, clinical applications, and limitations of current models. We focus our review on models of FCD type II that are based on genetic manipulations of the mTORC1 pathway (e.g., transgenic and IUE-based models) and also highlight other earlier models based on embryonic and neonatal acquired factors.
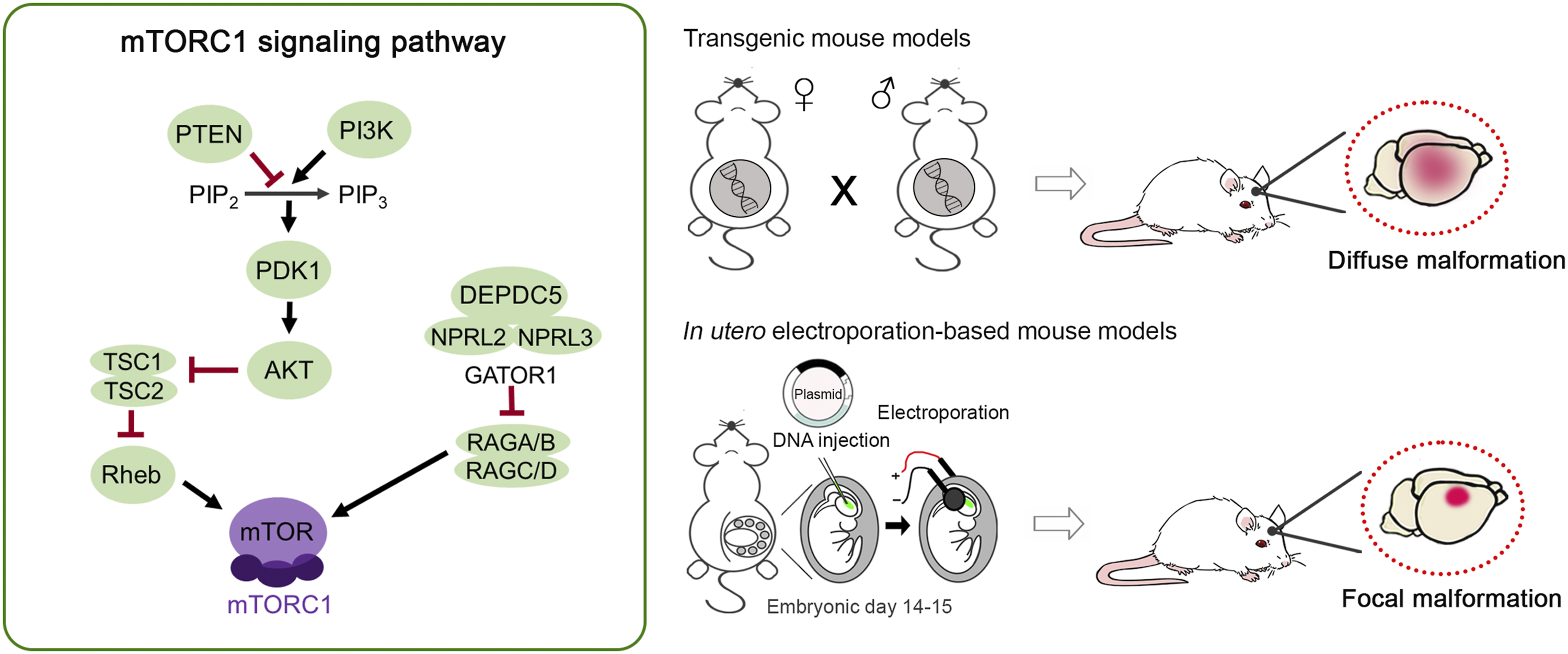
Animal models of focal cortical dysplasia (FCD) targeting the mTORC1 signaling pathway. The mTORC1 signaling pathway is a key regulator of neuron growth and development. Mutations in genes encoding regulators of the mTORC1 pathway, including MTOR, PIK3CA, PTEN, TSC1, TSC2, RHEB, DEPDC5, NPRL2, and NPRL3 have been identified as a cause of FCD type II. Based on this, many animal models of FCD type II have been developed by genetically targeting various components of the mTORC1 pathway, either through genetic engineering (transgenic mice) or in utero electroporation. Transgenic mice result in diffuse malformation while in utero electroporation-based models result in focal malformation. Both models recapitulate key phenotypes associated with FCD type II, including cortical disorganization, neuronal misplacement, presence of enlarged dysmorphic neurons, and spontaneous seizures.
Transgenic Animal Models
Most transgenic mouse models of FCD target single genes of the mTORC1 pathway to increase mTORC1 signaling, and they recapitulate major pathological features of FCD type II, including enlarged cortex, ectopic neuronal placement, neuronal cytomegaly, and epilepsy. Due to the histopathological similarities between FCD type II and cortical tubers in TSC, 5,6 many animal models of TSC have been interchangeably used to study FCD pathogenesis.
Common models of FCD type II include brain-specific conditional knockout of Pten, 14 -17 Tsc1 18 -24 , and Tsc2. 25,26 Mice with conditional knockout of Depdc5 27 or expression of Pik3ca 28 or mTOR 29 activating mutations have also been described. Different promoters have been used to knockout or knockin gene expression in various brain cells, including neural progenitor cells (and consequently all the cells in its lineage), excitatory neurons, interneurons, astrocytes, oligodendrocytes, and microglia. While the impact on neuronal physiology may differ, these manipulations share several common effects on neurons, regardless of their excitatory or inhibitory type, including increased mTORC1 signaling, impaired migration, and enlarged cell size. Knockout of Pten, 14 -17 Tsc1 18 -24 , Tsc2, 25,26 and Depdc5 27 or expression of Pik3ca 28 and mTOR 29 activating mutations in excitatory neurons (with or without expression in glia) lead to spontaneous seizures, while knockout of Pten 30 and Tsc1 31 in interneurons leads to altered EEG power and decreased seizure threshold on exposure to convulsant agents, respectively. Large, binucleated cells with enlarged vacuoles resembling balloon cells have been described in one of the transgenic models, indicating that this is a model of FCD type IIb. 22
Transgenic mouse lines are relatively easy to maintain in the laboratory and produce consistent pathology across animals and litters. The use of these mice has been key for establishing the role of mTORC1 signaling in cortical malformations and epilepsy. Conditional Pten, 14 -17 Tsc1, 21 -24,32,33 and Tsc2 26 knockout mice were originally used to demonstrate the anti-seizure effects of the mTOR inhibitor rapamycin on cortical malformation-related epilepsy, and they serve as established preclinical models to test novel therapeutics.
There are several limitations to using transgenic mouse models. In particular, multiple brain regions (including cortex, hippocampus, and cerebellum) and cell types are affected, making studies to elucidate the origin and pathogenesis of epilepsy challenging. To address this issue, exogenous injections of a Cre-expressing viral vector have been performed in Tsc1 floxed/floxed mice to achieve mosaic Tsc1 loss in a localized region similar to those observed in individuals with FCD and TSC. 34 Furthermore, many of the transgenic mouse models have severe pathological phenotypes and suffer from premature death (some as early as < postnatal day 21). 21,23,29,35 Despite having a well-characterized seizure phenotype, the seizure frequency is often low (<1 per day) 14,15,17,22,36 and highly variable between days and animals, and long-term monitoring is often needed to observe any treatment effects on the seizure phenotype.
In addition to mice, there are a few transgenic zebrafish models of cortical malformations and epilepsy, including Tsc1, 37 Tsc2, 38 and Depdc5 39 knockout zebrafish. These models recapitulate increased mTORC1 signaling, abnormal brain and neuron morphology and organization, and epileptiform activity, and they are particularly useful for high-throughput screening to identify new therapeutics for FCD-related epilepsy.
In Utero Electroporation-Based Animal Models
A major limitation of the transgenic models is the diffuse nature of the malformation across the cortex and other brain regions, lacking the focal localization of FCD lesions. In recent years, newer animal models of FCD based on in utero electroporation (IUE) to express mTORC1 pathway gene variants in the rodent cortex have emerged that better recapitulate the focal lesions of FCD. These are models of FCD type II and were developed in parallel with accumulating evidence supporting FCD type II pathology is caused by brain somatic mutations in genes along the mTORC1 pathway occurring in neural progenitor cells during embryonic development. 7,9,10
IUE is a technique that allows for in vivo gene manipulation in specific cell types in select regions of the developing brain. 40 With IUE, a plasmid DNA is introduced into the developing embryonic brain via microinjection into the ventricles and transfected by electrical pulses into a subpopulation of neural progenitor cells in the ventricular zone. All reported IUE-based models thus far target the progenitor cells of the cortical ventricular zone that generate excitatory pyramidal neurons. These neurons undergo migration and final differentiation in distinct cortical layers according to their birth date. Some of these progenitor cells will also differentiate into astrocytes and oligodendrocytes. 41 By controlling the timing, area, and direction of electroporation, a specific cortical layer and region can be targeted. The IUE-based models take advantage of this technique to create a localized malformation in the cortex that more precisely mimics the FCD type II pathology compared to the diffuse malformations in transgenic mice.
Current IUE-based animal models of FCD type II include the expression of gain-of-function mutations in mTOR or mTORC1 positive regulators (Pik3ca, Akt3, and Rheb) and loss-of-function manipulations (by CRISPR/Cas9-mediated knockout or shRNA-mediated knockdown systems) of the mTORC1 negative regulators (Pten, Tsc1, Tsc2, Stradα, Depdc5, and Nprl3) in wildtype rodents, mostly mice but also some rats. These models have been discussed in a previous review. 42 Many of these models use identified patient variants, thereby reproducing specific genetic molecular pathogenesis with high clinical relevance. In addition, there are IUE-based 2-hit models wherein a Cre recombinase plasmid is expressed by IUE in Tsc1 43 or Depdc5 44 floxed/mutant mice to better recapitulate the genetic mechanisms of some patients.
The existing IUE-based models target progenitor cells destined for either the somatosensory or medial prefrontal cortex and all lead to increased mTORC1 activation in excitatory pyramidal neurons. It is important to note that although IUE targets progenitor cells that give rise to both neurons and glial cells, episomal plasmids used in overexpression experiments will only be expressed in neurons due to a plasmid dilution effect that occurs in dividing cells. 43,45 In contrast, CRISPR/Cas9 plasmids used in knockout experiments will integrate into the genome of the electroporated progenitor cells and affect all cell types in the cell lineage (i.e., pyramidal neurons, astrocytes, and oligodendrocytes). 46 As with the transgenic models, IUE-based models recapitulate major FCDII phenotypes, including neuron migration defects and abnormal cortical lamination, enlarged dysmorphic neurons, and spontaneous seizures. 42 The presence or absence of balloon cells have not been well characterized in these models, and therefore, it is unclear whether they specifically model FCD type IIa or IIb.
A major advantage of the IUE-based models is that they provide region- and cell type-specificity and better recapitulate the focal nature of the FCD lesions. Moreover, by using IUE, newly identified patient variants can be introduced into the rodent cortex to study pathogenesis and mechanisms in a relatively simple manner and fast timeframe compared to creating transgenic lines. For unclear reasons, the IUE-based animal models tend to have a higher seizure frequency (∼3-10 per day) 47 -50 than the transgenic models, making them particularly advantageous for evaluating treatment effects since an impact on seizures would potentially be more readily detectable. The severity of the seizure phenotype can be titrated using different amounts of electroporated plasmids to address various questions. 50 Despite having a robust seizure phenotype, IUE animals are overall healthy and most have a normal lifespan. The IUE-based models have been particularly useful for identifying or validating potential molecular targets for the treatment of epilepsy in FCD type II. Some candidates that have emerged using IUE-based models of FCD type II include 4E-BP1, 51 eIF4E, 52 HCN4 53 Filamin A, 54 and adenosine kinase, 52 opening up opportunities for precision medicine approaches to treat epilepsy in FCD using gene or small molecule therapies. Additionally, the IUE-based models have also been used to test the anti-seizure effects of new or known drugs, such as rapamycin, 48,49,55 -57 PTI-125, 54 metformin, 52 and PD0325901. 58
One limitation to these models is that IUE is technically challenging and highly skill-dependent. New investigators often face low transfection efficiency, reproducibility issues, and low pup survival rates. While there will be some variability in terms of the electroporated region and transfection efficiency between animals, litters, and investigators, with practice, variability can be reduced, and pup survival rates may be as high as 100%.
Other Animal Models
Historically, MCD in animals was modeled by chemical or physical induction during early life to create cortical malformations. While these are not exclusively models of FCD, they share overlapping pathological phenotypes, such as laminar disorganization, cytomegalic neurons, and neuronal heterotopia, which have made them useful in studying FCD pathogenesis. These models have been the subjects of other reviews and are only briefly listed here. 59,60 These include: neonatal freeze lesion or in utero irradiation to generate microgyria and focal heterotopia, in utero exposure to methylazoxymethanol (MAM) acetate to reproduce periventricular nodular heterotopia, in utero exposure to carmustine 1-3-bis-chloroethyl-nitrosourea (BCNU) to induce laminar disorganization, heterotopias, and cytomegalic neurons, and neonatal exposure ibotenic acid to generate focal cortical malformations with microgyria, ulegyria, and periventricular nodular heterotopia. Both the freeze lesion (when performed at embryonic day 18) and in utero irradiation models are accompanied by spontaneous seizures although these are rare. The remaining models display enhanced cortical excitability and reduced seizure threshold. An adaptation of the MAM model has been performed by adding postnatal pilocarpine treatments, which led to a high recurrence of spontaneous seizures. 61 Since the discovery that mutations in mTORC1 pathway genes cause FCD type II, it is apparent that these models are not applicable for FCD type II. It is arguable whether they are representative models of FCD type I or III.
Conclusions and Future Directions
Animal models of FCD have been and continue to be invaluable tools for investigating mechanisms of epileptogenesis and novel treatments for epilepsy. Most are models of FCD type II, and these are transgenic and IUE-based models that target genes of the mTORC1 pathway and recapitulate, to various degrees, the cortical malformations and recurrent spontaneous seizures. Animal models of FCD type I are lacking and much needed. 13 These will become more feasible to develop as more knowledge on the etiology and molecular pathogenesis of FCD type I emerge. Animal models of FCD type III have been challenging to develop due to complicated and often contradictory definitions. 6 Better models could be developed once a clearer definition of FCD type III is established.
The presence of FCD malformations causes seizures but whether the source of epileptogenicity originates from within the FCD lesion or in the disrupted perilesional cortex remains an ongoing debate. Localizing seizures in the transgenic animal models is constrained by the diffuse nature of the malformations. Limited studies have attempted to determine the origin of seizures in the IUE-based models but the results are contradictory. 53,62 Thus, there is a need for studies to better localize and define the local circuits involved in FCD-related epileptogenesis. Another discussion with regards to the current FCD animal models is which cell types should be targeted. A recent study using single-cell sequencing of resected brain tissue from individuals with FCD type II demonstrated that somatic mutations in mTOR pathway genes can occur in dorsal telencephalic progenitors that generate both neuronal and non-neuronal cells or in neurons-specific progenitors. 9 In the former case, the somatic mutations were enriched in neurons compared to glial cells. 9 This is supported by another human genetics study showing that dysmorphic neurons and balloon cells in FCD type II are the main cells that carry the mTOR activating variants. 10 Although it is conceivable that these somatic mutations can also occur in progenitor cells of the ganglionic eminences that give rise to interneurons, this has yet to be described. In some of the animal models reviewed here, the genetic manipulations were performed in neural progenitor cells giving rise to both excitatory neurons and glial cells. In other models, only excitatory neurons were targeted, and this was shown to be sufficient for epileptogenesis. In contrast, selectively targeting interneurons did not result in seizures. 9,30,31 New insights into the cellular origins of the gene mutations will help refine the FCD type II animal models.
Overall, there is no “perfect” animal model as no single model recapitulates all the human phenotypes associated with FCD. Animal models should be selected based on the research questions that are being asked. One major limitation of using rodent models to study developmental cortical malformations is that these models lack certain human-specific developmental events (e.g., cortical expansion and gyrification) and cell types (e.g., outer radial glia). To this end, recent models of developmental cortical malformations based on patient-derived induced pluripotent stem cells and cerebral organoids have been developed. 63 -65 Complementary studies using animal models and human in vitro models will provide invaluable insights into the cellular mechanisms of cortical malformations and epilepsy.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institute of Neurological Disorders and Stroke (R01 NS111980) and Eunice Kennedy Shriver National Institute of Child Health and Human Development (F32 HD095567).