
Abstract
Chaudhary R, Albrecht S, Datunashvili M, Cerina M, Lüttjohann A, Han Y, Narayanan V, Chetkovich DM, Ruck T, Kuhlmann T, Pape HC, Meuth SG, Zobeiri M, Budde T. Cerebral Cortex. 2022 Jan 25;bhab491. doi:10.1093/cercor/bhab491. A consensus is yet to be reached regarding the exact prevalence of epileptic seizures or epilepsy in multiple sclerosis (MS). In addition, the underlying pathophysiological basis of the reciprocal interaction among neuroinflammation, demyelination, and epilepsy remains unclear. Therefore, a better understanding of cellular and network mechanisms linking these pathologies is needed. Cuprizone-induced general demyelination in rodents is a valuable model for studying MS pathologies. Here, we studied the relationship among epileptic activity, loss of myelin, and pro-inflammatory cytokines by inducing acute, generalized demyelination in a genetic mouse model of human absence epilepsy, C3H/HeJ mice. Both cellular and network mechanisms were studied using in vivo and in vitro electrophysiological techniques. We found that acute, generalized demyelination in C3H/HeJ mice resulted in a lower number of spike-wave discharges, increased cortical theta oscillations, and reduction of slow rhythmic intrathalamic burst activity. In addition, generalized demyelination resulted in a significant reduction in the amplitude of the hyperpolarization-activated inward current (Ih) in thalamic relay cells, which was accompanied by lower surface expression of hyperpolarization-activated, cyclic nucleotide-gated channels, and the phosphorylated form of TRIP8b (pS237-TRIP8b). We suggest that demyelination-related changes in thalamic Ih may be one of the factors defining the prevalence of seizures in MS.
Commentary
Neuroscience textbooks explain to us how the thalamus serves as a central hub that relays sensory information to the cortex. Often, these textbooks also describe how interconnected neural circuits between the thalamus and cortex serve as efficient propagators of seizures associated with the generalized epilepsies. But perhaps less appreciated are the numerous reports demonstrating that thalamic dysfunction is also observed in multiple sclerosis (MS). Indeed, lesions are often clearly evident in thalamic structures within the brains of MS patients and may contribute to neuropathic pain and fatigue associated with the degenerative disease. But how such lesions affect generalized seizures remains unknown. Authors of a recent report in Cerebral Cortex now provide important and surprising insights into the interaction between MS and generalized seizures. 1
The first medical description of MS was likely provided in 1840 by William McKenzie, and then in 1868 Jean-Martin Charcot provided the first comprehensive account of the sclerotic lesions – he called the lesions “sclerose en plaques” – we now associate with the degenerative disease. 2 We now understand that these lesions represent pockets of demyelinated axons. Without myelin, axons are unable to rapidly propagate action potentials, effectively reducing the capability of neurons to efficiently propagate the most common and important electrical signals produced by neurons. The general consensus is that MS results when inflammatory CD8+ T cells attack oligodendrocytes, glial cells responsible for making myelin. 3 For this reason, MS has historically been referred to as a white matter disease. However, ample evidence demonstrates that neuronal cell bodies, the so-called gray matter of the brain, also lay victim to the immune system’s T lymphocytes. MS lesions are found throughout the central nervous system, including the cerebellum, brainstem and spinal cord, and a patient’s symptoms largely reflect damage to the affected structures.
As MS ultimately affects the electrical signaling produced by neurons, it is perhaps not too surprising that the autoimmune disease finds intersectional points with epilepsy, a disorder defined by periodic bouts of excessive neuronal signaling. Indeed, the incidence of epilepsy among MS patients is between three- and 6-fold greater that in the otherwise healthy population.
4
Moreover, the first indication of MS often comes in the form of a seizure. Thus, the two disorders appear tightly linked, leading some to speculate that the relationship between MS and epilepsy is “more than coincidental”.
4
Whereas the evidence linking the two disorders largely comes from clinical reports, results from the few existing rodent studies suggest that demyelination can cause epilepsy. For example, EEG recordings reveal that mice produce spontaneous spikes after 12 weeks of eating cuprizone, a copper chelating agent that causes demyelination (Figure 1A).
5
Cuprizone-treated mice are also more susceptible to stress-induced generalized tonic-clonic seizures. Histological examination reveals that the hippocampi of treated mice are extensively demyelinated. Notably, whereas cuprizone treatment recapitulates MS-associated demyelination, the decades-old model does not involve an autoimmune component synonymous with the disorder.
6
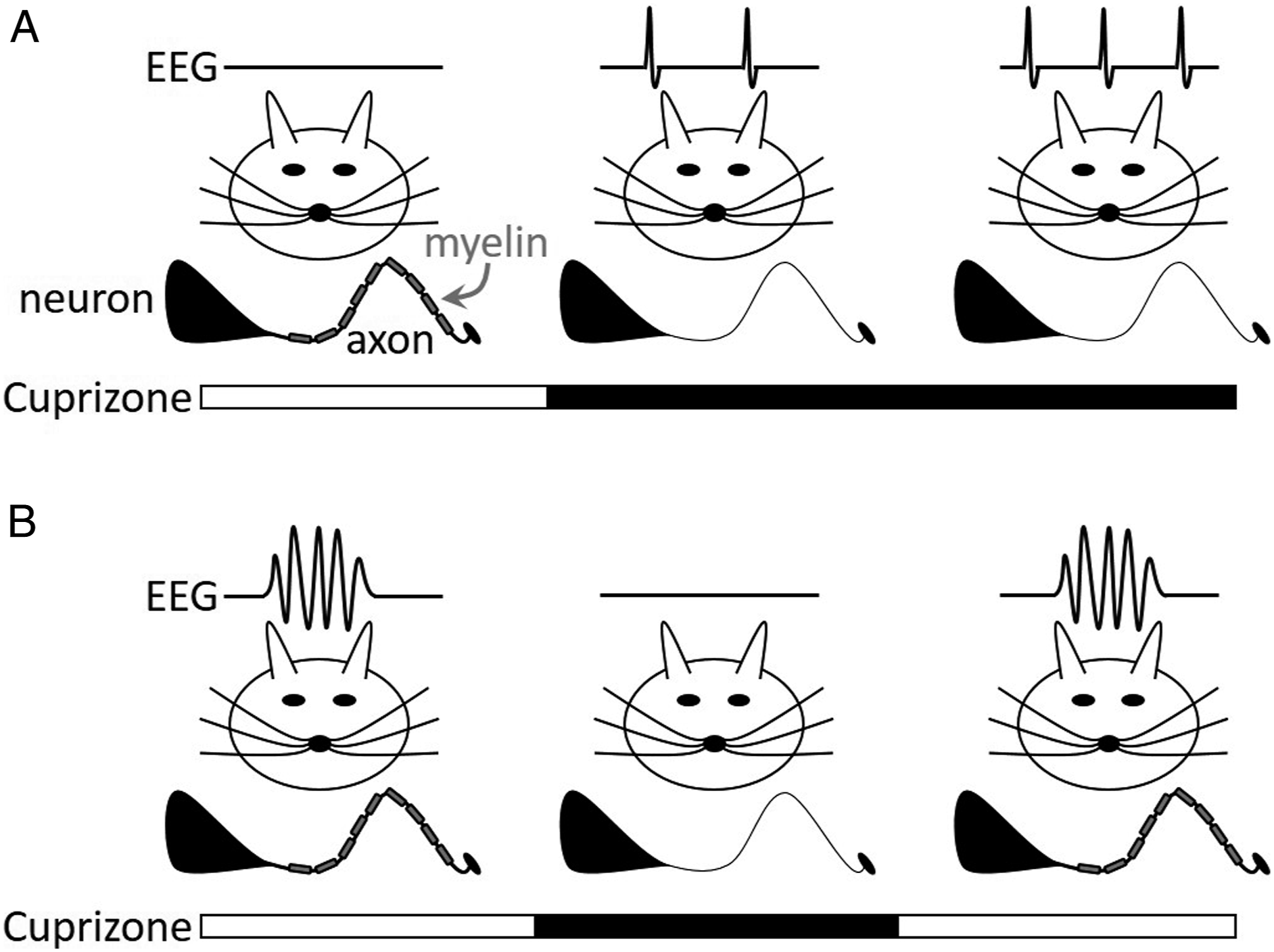
But what happens when demyelination is induced in mice already producing spontaneous seizures? This question was recently addressed by Chaudhary et al, 1 and the results are quite surprising. Rather than exacerbating seizures, cuprizone treatment of already epileptic mice reduced seizures.
To evaluate the effects of demyelination on epileptic mice, Chaudhary et al. treated the Gria4 mouse, 7 an established model of generalized absence (i.e., petit mal) seizures, with cuprizone for 5 weeks. At the 5-weeks mark, cuprizone treatment was suspended to enable remyelination, a process that is rapidly mobilized but may take a few weeks to fully complete. 6 Whereas age-matched, control Gria4 mice produced roughly 8 spontaneous spike-wave (i.e., absence) seizures per hour, fully demyelinated mice produced between 2-3 seizures per hour (Figure 1B). During the 4-week, post-cuprizone remyelination phase, the number of spike-wave seizures produced by treated Gria4 mice gradually increased. Although systemic demyelination undoubtedly has extensive actions throughout the CNS, the authors also demonstrate that altered cellular and network excitability in thalamic structures appears sufficient to explain reduced seizure expression in demyelinated Gria4 mice. To this end, rhythmic, neuronal bursting activity generated in acute brain slices containing only the somatosensory and reticular thalamus was evaluated; bursting was evoked with electrical stimulation of the axons belonging to thalamocortical and corticothalamic neurons. Consistent with the effects on spontaneous seizures, fewer bursts were evoked in fully demyelinated slices, an effect that gradually waned during the 4-week remyelination phase.
With the stage set, the authors then attempt to resolve the cellular mechanisms accounting for reduced burst excitability in demyelinated thalamic slices. The conclusion derived after a series of electrophysiological and immunohistochemical experiments is that demyelination downregulates a mixed cation current mediated by hyperpolarization-activated cyclic nucleotide-gated HCN channels. Responsible for pacemaker activity in thalamocortical neurons, HCN channels are integral for the generation of both healthy and seizure-related thalamic oscillations. 8 As thalamocortical neurons in demyelinated brains produce a reduced level of HCN current, the hypothesis goes, the capability of such neurons to support spike-wave seizures is likewise diminished.
But how exactly does a reduction in the conductive myelin sheath surrounding axons downregulate HCN channel density in thalamocortical neurons? The answer, of course, is complex and incompletely understood. Rapidly demyelinating the brain produces many effects, including an inflammatory response that includes a robust activation of astrocytes and microglia. Nonetheless, Chaudhary et al. provide data consistent with one hypothesis: cytokines released by microglia ultimately drive down HCN channel expression. Activated microglia release the type I interferons (IFN) IFNα and IFNβ in animal models of MS, 9 and the authors show that both thalamocortical HCN currents and thalamocortical neuron bursting propensity are modulated by the two interferons. Curiously, however, IFNα and IFNβ exert opposing actions. HCN currents and bursting are downregulated with exogenous IFNα application, whereas both properties are upregulated by IFNβ application. Thus, a complex model emerges. The authors speculate that microglia release of IFNα in the demyelinated thalamus reduces HCN currents that ultimately support absence seizures. During the remyelination phase, microglia release of IFNβ restores thalamocortical neuron HCN currents and, thus, restores epileptogenicity.
The model is speculative, and many questions remain. First of all, it remains unclear if a human counterpart exists wherein a patient with an existing diagnosis of absence epilepsy develops MS. While such patients very likely exist, absence seizures – at least those associated with typical childhood absence epilepsy – usually remit in the early 20s, whereas the age of onset for MS is generally between 20 and 40 years of age. Thus, such patients are likely uncommon. More likely, MS patients may have preexisting focal epilepsy or other types of generalized seizures, but the relationship between MS and other seizures was not addressed in the current study. Finally, the most common situation would be the emergence of seizures in MS patients with no pre-existing history of epilepsy. In this context, the current study offers no explanation for the apparent paradox that cuprizone-induced demyelination decreases seizures in the absence epilepsy model, but triggers seizures in non-epileptic mice in prior studies. Nevertheless, the author’s model underscores the environmental complexity in which glial cells and neurons collectively regulate brain function.