
Abstract
Lee CH, Le JT, Ballester-Rosado CJ, et al. Ann Neurol. 2020;89(2):226-241. doi:10.1002/ana.25935 Epileptic spasms are a hallmark of severe seizure disorders. The neurophysiological mechanisms and the neuronal circuit(s) that generate these seizures are unresolved and are the focus of studies reported here. In the tetrodotoxin model, we used 16-channel microarrays and microwires to record electrophysiological activity in neocortex and thalamus during spasms. Chemogenetic activation was used to examine the role of neocortical pyramidal cells in generating spasms. Comparisons were made to recordings from infantile spasm patients. Current source density and simultaneous multiunit activity analyses indicate that the ictal events of spasms are initiated in infragranular cortical layers. A dramatic pause of neuronal activity was recorded immediately prior to the onset of spasms. This preictal pause is shown to share many features with the down states of slow-wave sleep. In addition, the ensuing interictal up states of slow-wave rhythms are more intense in epileptic than control animals and occasionally appear sufficient to initiate spasms. Chemogenetic activation of neocortical pyramidal cells supported these observations, as it increased slow oscillations and spasm numbers and clustering. Recordings also revealed a ramp-up in the number of neocortical slow oscillations preceding spasms, which was also observed in infantile spasm patients. Our findings provide evidence that epileptic spasms can arise from the neocortex and reveal a previously unappreciated interplay between brain state physiology and spasm generation. The identification of neocortical up states as a mechanism capable of initiating epileptic spasms will likely provide new targets for interventional therapies.Objective:
Methods:
Results:
Interpretation:
Commentary
Infantile spasms (IS), and the analogous syndrome in which spasms begin after 1 year of age (epileptic spasms, ES), is a severe developmental epilepsy syndrome (an epileptic encephalopathy) that has haunted clinicians and neuroscientists for decades. The unique seizure semiology (flexion/extension spasms), characteristic electroencephalographic (EEG) features (hypsarrhythmia), and wide-ranging etiologies of IS and ES (well over 250 acquired and genetic causes have been identified) suggest that some common pathophysiological processes drive seizure production and the encephalopathy. Yet, numerous questions remain about the underlying molecular and cellular mechanisms. Infantile spasms and ES are often uniquely responsive to unconventional antiseizure treatments, namely adrenocorticotrophic hormone and corticosteroids, and to some extent, vigabatrin or the ketogenic diet. Nevertheless, those and other treatments are often not effective and carry significant side effects, while the long-term neurodevelopmental and cognitive outcomes of IS and ES remain poor. Several animal models have been developed as an attempt to address some of these knowledge gaps, but no model fully reproduces the human syndrome. 1 This situation is not surprising, given the vast species differences in cortical structure and physiology. Each animal model is poised to answer certain questions about spasms, but none of them will reveal all of the mysteries. Two questions seem eminently amenable to an experimental approach using animal models: (1) where in the brain do spasms originate (cortical, subcortical, both, neither), a question that has been long debated, and (2) what is the physiological basis of the unique EEG signatures in spasms, namely, interictal hypsarrhythmia and ictal electrodecrement? Further elucidation of either of these questions could inform novel approaches to treatment.
More than a decade ago, Lee and colleagues developed an animal model of ES by injection of the sodium channel blocker tetrodotoxin (TTX) into the brains of infant rats, using osmotic mini-pumps. 2 This model mimics spasms that have an acquired etiology. In rat pups (P10-12), when TTX is applied directly into the cortex of one hemisphere, IS-like flexion seizures occur about a week later and continue for weeks, even after the TTX injection is stopped. It is unknown how TTX exposure leads to the spasms, but TTX abrogates local neuronal firing and chronically suppresses neural activity during a specific developmental window, which engenders hyperexcitability, perhaps by “desynchronizing” the neuronal activity. 3 The existence of a latent period between TTX injection and spasms occurrence mimics the latent period in humans, during which it is presumed that epileptogenesis is taking place. Perhaps the most striking feature of the TTX model is the similarity of interictal EEG findings (hypsarrhythmia) and ictal EEG signature (high-voltage synchronous slow wave followed by electrodecremental response) in human infants and rat pups. Swann’s group has carefully characterized these EEG changes in their model. They previously showed that spasms can arise from cortex both ipsilateral and contralateral to the site of TTX administration, though the contralateral side seems especially prone to generate spasms. 2,4,5 High-frequency oscillations (HFOs, 250-600 Hz) were seen during ictal events occurring contralateral to the side of TTX injection. 4,5 High-frequency oscillations are accompanied by a burst of multiunit activity (MUA) 5 and may correlate with sites of ictogenesis. 6 The group is now pushing the frontier forward by examining the site of spasms onset and the mechanisms involved in spasms generation.
The present article asks whether contralateral cortex can generate ES, and if so, which cortical layers are involved. 7 The authors utilize a variety of approaches to evaluate the hypothesis that the contralateral cortex can generate ES. First, using current source density analysis, they show that the “heralding” ictal slow wave, representing the initial electrographic manifestation of the ictal phase (spasm), is generated in infragranular layers (layers V, VI) of contralateral cortex (Figure 1). Layers V and VI harbor large current sinks and represent dense excitatory networks; these results suggest that local cortical circuits generate spasms. Figure 1 shows a hypothetical EEG wave consisting of interictal high-voltage irregular slow waves with intermixed sharp waves (hypsarrhythmia), interrupted by the ictal slow wave leading into a clinical spasm, during which the EEG activity “decrements” for up to several seconds while the spasm occurs. The ictal slow wave is accompanied by the appearance of HFOs and MUAs.
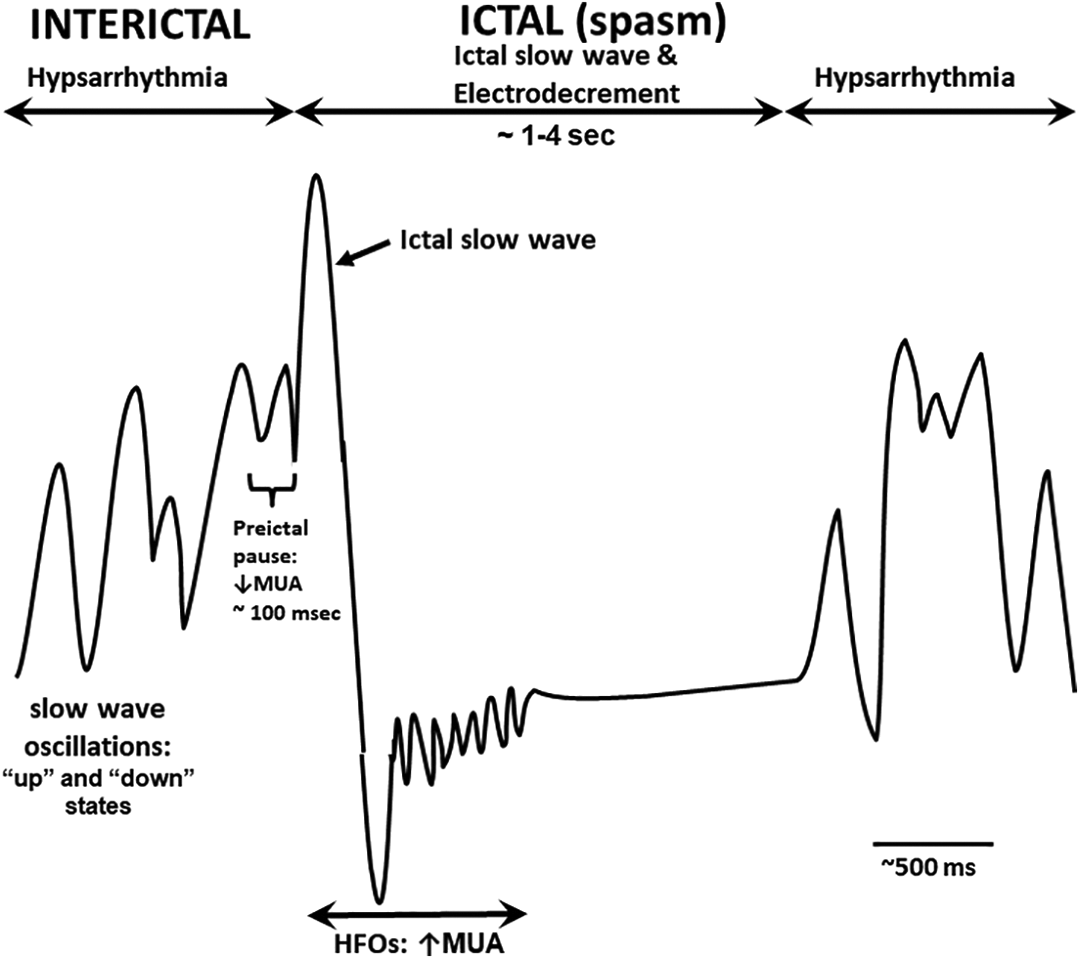
Schematic showing one channel of a hypothetical electroencephalographic (EEG) waveform in infantile spasms, including interictal hypsarrhythmia and ictal electrodecrement. Hypsarrhythmia is characterized by high voltage, irregular slow waves with superimposed sharp waves and spikes. Slow-wave oscillations resembling the cortical up and down states of slow-wave sleep occur during hypsarrhythmia; prior to onset of a spasm, slow-wave oscillations increase. 7 For approximately 100 msec prior to the ictal spasm, multiunit activity (MUA) decreases dramatically in infragranular layers (cortical layers V and VI) and is referred to as the called the preictal pause. The ictal phase (spasm) is heralded by an initial high-voltage slow wave (ictal slow wave) that typically precedes the ictal electrodecrement. The ictal slow wave and early part of the electrodecrement are characterized by high-frequency oscillations (HFOs) thought to be mediated by gap junctions. 8 Once the spasm ends, the electrodecrement ceases and hypsarrhythmia resumes.
A surprising finding from the article was that just prior to spasm onset, MUA activity ceased for about 100 msec—called the “preictal pause” 7 (see Figure 1). The cessation of neuronal activity during the preictal pause suggests that local neuronal activity is inhibited, similar to what is seen in the so-called “down state” of cortical activity accompanying slow-wave sleep. 9 Since the preictal pause resembles the cortical down state, the authors next examined whether the ictal phase is initiated by a subsequent cortical “up state.” Indeed, as the ES ictus ensues, there is a marked increase in pyramidal cell and interneuron firing, just as seen in the up state of sleep. Rats with ES had many more oscillations of up and down states in the minutes preceding a spasm than did rats without ES. These findings implicate an interplay between brain state mechanisms and spasm generation, an observation that is not surprising given the well-recognized relationship between sleep state, hypsarrhythmia, and spasms. The authors include some supportive human EEG data depicting preictal oscillations in local field potentials, similar to those seen in epileptic rats.
To confirm that cortical pyramidal cells participate in spasm generation, the authors used Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) to directly activate those cells. 10,11 They found that chemoactivation of infragranular neurons using DREADDs increased the number of interictal slow-wave oscillations (up and down states) just prior to onset of a spasm as well as the number of spasms and their clustering. These results support the notion that spasms originate in neocortex and provide a powerful experimental approach to “control” neuronal firing and hence dissect the mechanisms involved in spasms. This idea would be further supported if it could be shown that activation of inhibitory pathways blocked spasms.
This article provides some significant advances in our understanding of the physiology of ES, particularly their generation in the infragranular cortical layers and the relationship between cortical up and down states of slow-wave sleep and spasm generation. It is well known that IS occur most frequently as an infant awakens from sleep or becomes drowsy. In other epilepsies as well, there is a marked relationship to sleep state. Therefore, it is not surprising that modulation of sleep can alter the occurrence of IS, opening a possible avenue to specific intervention. 12,13 As mentioned above, animal models can enhance our understanding of human ES, and this article also provides some direct evidence of the similarities in those 2 species. It would be important to show that similar mechanisms were operative in other IS models, before generalizing these results. Much more work needs to be done to both parse out the pathophysiological mechanisms of IS. Among the remaining questions are the mechanisms underlying the specific time windows of susceptibility to IS and ES, features of the latent period between the initiating insult and spasms occurrence, and the membrane and cellular correlates underlying interictal and ictal phases. Recent work addresses ion channel and neurotransmitter dysfunction, among other cellular mechanisms, underlying the EEG patterns of IS. For example, in a cortical slice model, layer V bursting neurons demonstrate prolonged depolarizing plateaus during the ictal electrodecrement, suggesting that neurons may not actually be silent during the decrement, but rather display pathologically increased excitability, 8 not unlike that recorded in the TTX model. Many of these questions can be approached using animal models of IS, hopefully leading to the emergence of more efficacious therapies.