
Abstract
Perineuronal nets (PNNs) are condensed extracellular matrix (ECM) assemblies of polyanionic chondroitin sulfate proteoglycans, hyaluronan, and tenascins that primarily wrap around GABAergic parvalbumin (PV) interneurons. During development, PNN formation terminates the critical period of neuroplasticity, a process that can be reversed by experimental disruption of PNNs. Perineuronal nets also regulate the intrinsic properties of the enclosed PV neurons thereby maintaining their inhibitory activity. Recent studies have implicated PNNs in central nervous system diseases as well as PV neuron dysfunction; consequently, they have further been associated with altered inhibition, particularly in the genesis of epilepsy. A wide range of seizure presentations in human and rodent models exhibit ECM remodeling with PNN disruption due to elevated protease activity. Inhibition of PNN proteolysis reduces seizure activity suggesting that PNN degrading enzymes may be potential novel therapeutic targets.
Introduction
The extracellular space (ECS) between neurons and glial cells is occupied by a heterogeneous ground substance or extracellular matrix (ECM) that spans from a diffused and amorphous interstitial matrix to highly organized lattice-like neuronal ensheathments called perineuronal nets (PNNs). Perineuronal nets encapsulate the cell body, dendrites, and axon-initial segment, primarily of parvalbumin (PV)-expressing GABAergic neurons. They are found in many brain regions including cerebral cortex, hippocampus, and amygdala and have been documented in several mammals including rodent, sheep, monkey, and human.1,2
Although Camilo Golgi discovered PNNs already in the late 19th century, the understanding of their structure and function started surfacing only a century later. 1 Indeed, much of our detailed knowledge on the fundamental functions of PNNs in health and disease was obtained in the past 30 years. By and large, these studies suggest that PNNs are important regulators of neuronal plasticity. Perforations in the PNN lattice contain synapses and developmental deposition of PNNs is suggested to lock synapses in place, thereby preventing further plasticity. This explains, for example, the establishment of ocular dominance during development of the visual system 3 but has also been suggested to serve as a memory engram for very long-term memories. 4 However, recent studies suggest that PNNs are quite dynamic and are the substrate for a number of proteolytic enzymes including matrix metalloproteinases (MMPs), a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTs), and tissue plasminogen activator (tPA). Remodeling of PNNs is particularly important in the context of injury and disease and PNNs are now implicated in schizophrenia, traumatic brain injury, Alzheimer disease, and epilepsy.5-8,9-12 In this short review, we summarize current knowledge on the structure and function of PNNs and discuss a potential role for PNN dysfunction as a contributor to acquired epilepsy.
Structure and Formation of PNNs
Perineuronal nets are ternary structures of membrane-bound hyaluronic acid synthase (HAS)-associated hyaluronic acid or hyaluronan (HA) chains on which several chondroitin sulfate proteoglycans (CSPGs) or lecticans such as aggrecan, brevican, versican, neurocan, and so on 13 are covalently bound via link proteins such as hyaluronic acid and proteoglycan link proteins (HAPLNs; Figure 1B). Perineuronal nets can be readily identified by staining with antibodies to any of its constituents such as chondroitin-sulfate, aggrecan, tenascin, or HA, yet the most commonly used stain is the plant lectin wisteria floribunda agglutinin (WFA; Figure 1A).15,16 Besides the structural role, CSPGs and HA interact with membrane receptors such as CD44, leucocyte common antigen-related receptor, receptor type protein tyrosine phosphatases σ, Nogo receptors (NgR1, R3), epidermal growth factor receptor, integrins and so on and modulate several intracellular signaling pathways. 17
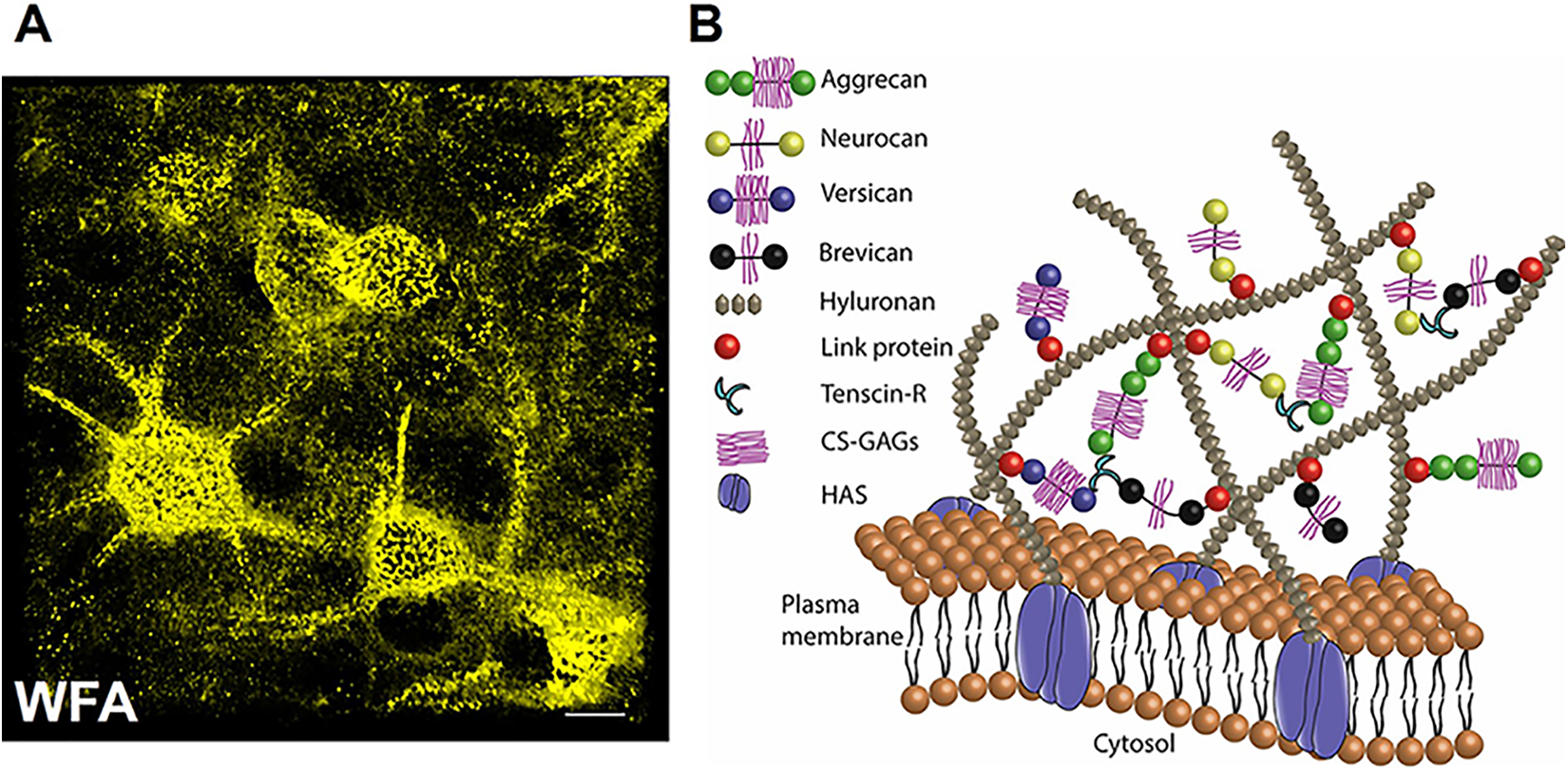
Perineuronal nets in the central nervous system. A, Immunohistochemical staining with fluorescently labeled WFA showing perineuronal nets in mouse cerebral cortex. Perineuronal nets surround cell body axon initial segments and dendrites (scale 10 µm). B, Schematics of the structural organization of PNN constituents on the PV neuron membrane. Long chains of HAS-associated HA are connected to the lecticans aggrecan, neurocan, brevican, and versican via link proteins. This multimolecular complex is further strengthened by tenascins, especially TnR, which crosslink lecticans, link proteins and HA to give rise to a lattice-like appearance. The sulfated proteoglycans turn PNNs into a sphere with a high density of stationary negative charges, which, in combination with the polarized groups of proteoglycans maintain ionic homeostasis and hydration capacity. 14 HA indicates hyaluronic acid/hyaluronan; HAS, hyaluronic acid synthase; PNNs, perineuronal nets; PV, parvalbumin; TnR, tenascin-R; WFA, Wisteria floribunda agglutinin.
The ECM molecules (Table 1) start assembling into PNNs in rodent brain in the second-third postnatal week and fully formed PNNs are seen by 5 to 6 weeks of age. 43 In human brain, PNNs appear as early as 2 months and show fully formed mature morphology around 8 years of age. 44 Brain activity is critical for the developmental PNN formation, and diminished brain activity upon sensory deprivation attenuates PNN expression. 6 Interestingly, the activity-dependent PNN formation occurs only during postnatal development, and sensory deprivation in adults affects neither PNN expression nor plasticity.3,9
Extracellular Molecules and ECM Remodeling Proteases in Epilepsy.
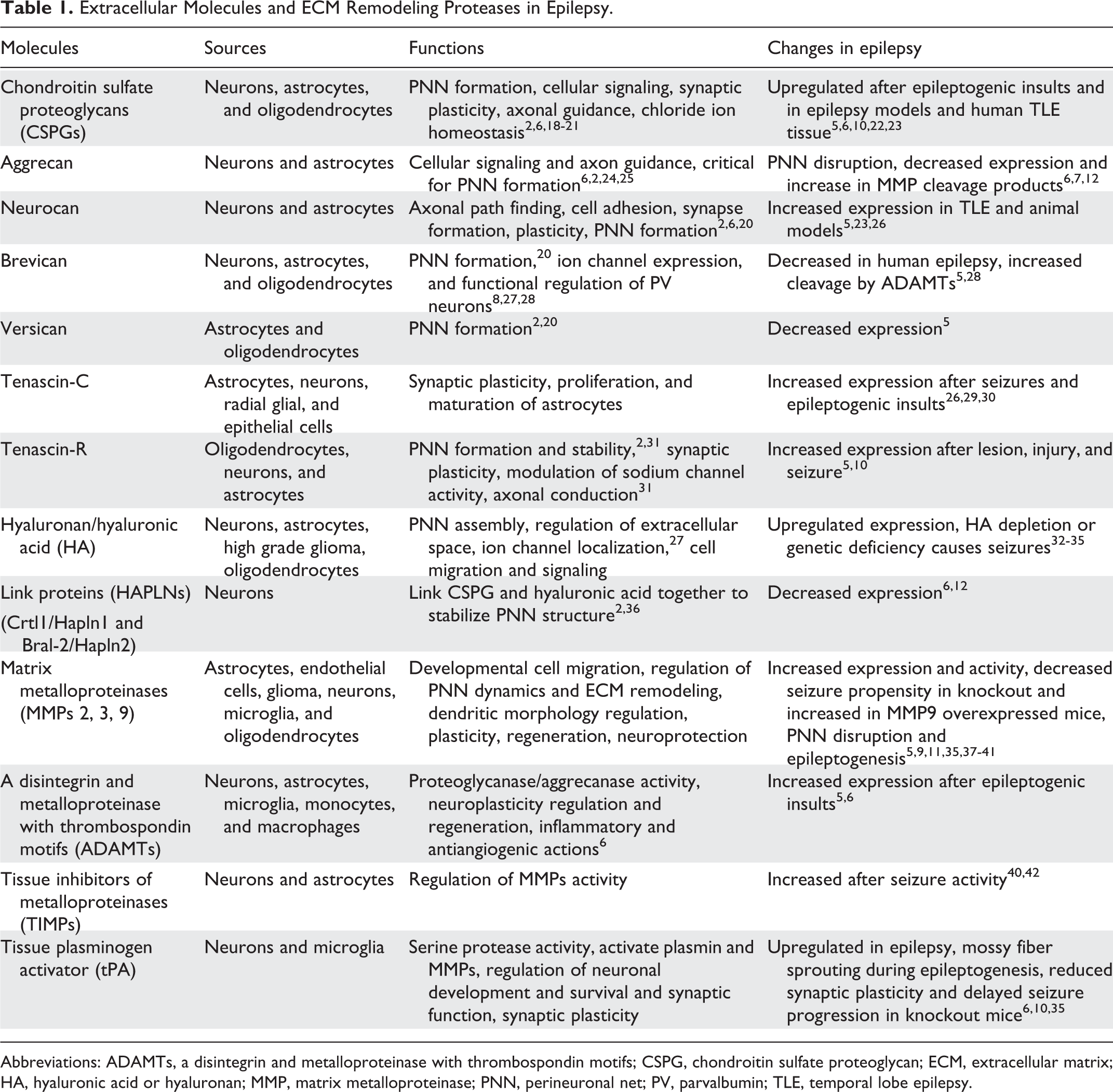
Abbreviations: ADAMTs, a disintegrin and metalloproteinase with thrombospondin motifs; CSPG, chondroitin sulfate proteoglycan; ECM, extracellular matrix; HA, hyaluronic acid or hyaluronan; MMP, matrix metalloproteinase; PNN, perineuronal net; PV, parvalbumin; TLE, temporal lobe epilepsy.
Spatiotemporal variation in the sulfation pattern of CSPG critically determines the plasticity permissive nature of the ECM or PNN during development and disease. Typically, CSPGs can be monosulfated at 4 (C4S) or 6 (C6S) carbon positions or bisulfated at 2, 6 or 4, 6 positions. C6S is permissive to axonal growth, regeneration, and plasticity, whereas C4S is inhibitory. 9 Owing to a high C6S expression, hence a low C4S/C6S ratio, developmentally immature brain without fully formed PNNs is permissive to neuroplasticity. As the brain matures, the sulfation pattern culminates in a higher C4S/C6S ratio in the adult brain and terminates critical period plasticity; additionally it also favors maturation of the PNN.18,19,36 Surprisingly, diseased states, especially epilepsy exhibit altered sulfation patterns 45 as well as maladaptive plasticity. 37
Functions of PNN
During development, several brain regions including the visual and barrel cortices undergo a characteristic critical period of heightened neuronal plasticity, during which sensory inputs drive synaptogenesis in an activity-dependent manner and cause loss of synapses which lack sufficient activity. 46 Interestingly, maturation of PV-expressing GABAergic interneurons triggers the onset of the critical period, and the appearance of mature PNNs around them ends the critical period, leading to the idea that PNNs, once formed lock synaptic contacts in place.3,9,46,47 This idea is further supported by the fact that preventing PNN maturation prolongs the critical period of plasticity beyond the typical developmental window, and PNN disruption beyond the critical period reinstates plasticity.3,9,24,36 Such experimental manipulation of PNNs can be achieved by genetically eliminating PNN constituents or by using enzymes that nonspecifically cleave the PNN proteoglycans20,24,48 including chondroitinase ABC (ChABC) or hyaluronidase (Hyase).
Disruption of PNNs causes several changes at cellular and molecular levels that shed some light on the potential mechanisms by which PNNs influence synaptic plasticity. For example, hippocampal area CA2 is normally resistant to plasticity; however, disruption of PNNs can generate synaptic potentiation and increase excitatory postsynaptic activity in CA2 neurons. 49 At hippocampal CA3-CA1 synapses, PNN digestion impairs long-term potentiation (LTP) by altering L type voltage-dependent calcium channel activity. 27 Perineuronal net digestion also increases γ activity 50 as well as the sharp wave ripples in CA1 stratum radiatum. 51 In addition, PNN depletion changes dendritic spine dynamics, alters synaptic contacts on PV neurons, and modulates expression of ion channels and receptors thereby promoting synaptic plasticity; see review of Fawcett et al 9 for details.
Several functions of the ECM appear to be primarily dependent on its specific constituents including CSPGs, HA, and tenascins, which are present in the PNNs as well as in the diffused interstitial matrix, however with a different density. For example, during prenatal central nervous system (CNS) development when PNNs are not yet formed, diffused interstitial CSPGs act as an axon guidance cue to prevent axonal movement toward nontarget areas, and CSPG depletion disrupts axonal guidance.25,52 Interestingly, upregulation of CSPGs after injury is primarily an obstacle in the regeneration of damaged axons, and ChABC treatment significantly improves the growth, regeneration, and functional recovery after injury. 53 Perineuronal nets compartmentalize certain membrane proteins and ion channels at a subcellular level, thereby directly affecting PV neuron activity. For example, condensed HA restricts the lateral movement of AMPA receptors (AMPAR) to prevent their exchange between synaptic and extrasynaptic sites. 54 Perisynaptic HA also regulates the expression of dendritic calcium channels to facilitate their use-dependent synaptic plasticity. 27 Brevican, a CSPG of the PNN, dynamically regulates the activity of enclosed PV neurons by controlling the localization of potassium channels and AMPARs on the membrane. Experimental manipulations, such as enriched environment rearing, can directly affect the brevican level to modulate PV neuron activity via altering potassium channel and AMPAR localization. 28 The high-density sulfated proteoglycans of the PNN attract and retain neuromodulatory molecules, including Semaphorin A, Narp, Otx2, and Reelin, which are critical for the functional maturation of PV neurons. 9 Parvalbumin neuron’s fast spiking can also be regulated by the PNN assembly as discussed in detail in the next section.
Role of PNNs in Epilepsy
Epilepsy is a nervous system disorder of spontaneous and recurrent seizures predominantly caused by an imbalance of excitation and inhibition (E-I). Since PNNs determine the excitability of PV interneurons, they indirectly control GABAergic inhibition. This brings PNNs to a central place to influence the delicate balance of E-I.
Perineuronal nets-expressing PV neurons possess an extraordinary spiking frequency (100-800 Hz); 55 hence they are called fast-spiking neurons, and recent studies by us 8 and others28,56 suggest that the presence of PNNs aids the fast-spiking of PV neurons. We 8 and more recently others 57 have shown that PNN assembly reduces the membrane capacitance of their enclosed PV neurons which in turn allows cells to exceed the spike frequency otherwise limited by their intrinsic membrane capacity.
In addition, the role of PNNs in synaptic plasticity and regulation of ion channel function are additional ways in which PNNs are potentially associated with epilepsy. Indeed, emerging evidence supports the idea that ECM remodeling and PNN disruption causes PV neuron dysfunction and maladaptive plasticity, which can shape epileptogenesis as well as tip the E-I balance to generate a seizure- as described in more detail below.
The condensed form of ECM as PNN is unique to the PV neurons; however, the diffused and amorphous interstitial matrix of similar molecular composition is omnipresent. Interestingly, both of these ECM forms are altered in epilepsy and potentially exert different outcomes. Biochemical and immunohistochemical studies on animal models and human temporal lobe epilepsy tissues show not only an abnormal expression of the individual ECM molecules including CSPG, heparin sulfate proteoglycan (HSPG), HA, aggrecan, and brevican,7,12,28,22,32,58,59 but also a disruption of the PNN’s structural integrity7,8,22 and often reduced density of PNNs and PV neurons.8,37 However, acquired epilepsies triggered by brain insults such as trauma, stroke, or brain tumors present more convincing evidence of an epileptogenic outcome of ECM remodeling and PNN disruption. The CNS injuries highly upregulate several CSPGs and HSPGs and occasionally change their sulfation pattern by overexpressing sulfotransferases. 45 In addition, low numerical density and structurally deficient PNNs are common after epileptogenic insults.5,22,59 Genomic and proteomic analyses of diverse epileptogenic injuries also confirm the upregulation of ECM-remodeling genes and proteins.5,22 In glioma-associated epilepsy, we8,10 and more recently others 60 have reported a reduced density of PNNs and PV neurons, as well as disrupted structural integrity of remaining PNNs due to glioma-released MMPs. This reduces the overall inhibition via altering the spiking properties of PV interneurons, and increases the overall seizure propensity. Interestingly, PNN disruption also characterizes several neurodevelopmental and neurodegenerative disorders in which seizures are comorbid; however, their involvement in core pathologies and seizure generation remains to be established. 61
The above studies suggest a paradox regarding ECM expression, wherein the individual ECM components are generally upregulated but the ECM composing the condensed PNNs is disrupted. However, ECM upregulation and PNN disruption seem to be through different mechanisms. Extracellular matrix upregulation is credited to reactive glial cells, primarily astrocytes, that release excessive ECM molecules as a generic response to CNS injury 5 which to a certain extent is confined to the injury site or the glial scar.45,53 On the other hand, PNN cleavage is primarily associated with upregulated proteases such as MMPs, ADAMTs, and tPA and to a lesser extent with the lower expression of the PNN-assembling molecules such as HAPLN, and MMP inhibitors such as tissue inhibitors of metalloproteinases.6,12,22 Interestingly, reactive glial cells, primarily astrocytes, appear to be the major sources of MMPs 62 and preventing MMP activity attenuates neuronal hyperactivity (see review of Kim et al 5 for details and Box 1).
Another line of evidence supporting the role of ECM remodeling and PNN disruption in seizure causality comes from studies showing spontaneous seizure-like activity or increased seizure susceptibility upon ECM and PNN deficiency in otherwise normal brains. 33 The best example is the HAS3 knockout mouse, in which HA deficiency causes spontaneous seizures due to altered ionic homeostasis and ephaptic interactions. 34 Tenascin-R (TnR) and tenascin-C (TnC) knockouts similarly show increased seizure susceptibility, 29 reduced perisomatic inhibition, increased excitatory transmission, and impaired LTP. 69 A quadruple knockout of TnC, TnR, neurocan, and brevican shows impaired PNN assembly to coincide with increased excitation and reduced inhibition. 70 However, whether recently developed aggrecan-deficient mice that lack WFA-expressing PNNs show any seizure activity is not known. 24 In addition to genetic approaches, enzymatic degradation of PNNs by ChABC or hyase has been shown to cause spontaneous seizures or epileptiform-like activity, 71 lower seizure threshold or latency of epileptiform activity, 8 and aggravated seizures.7,33,72
Matrix metalloproteinases: master regulator of PNN dynamics.
Extracellular matrix remodeling in the central nervous system (CNS) is primarily attributed to matrix metalloproteinases (MMPs), which are extracellular proteases produced by neurons, astrocytes, and oligodendrocytes that cleave proteins, proteoglycans, growth factors, and membrane proteins. 5 MMPs contain 25 members in their family, 3 of which are found in CNS and are known to cleave proteoglycans.6,7 A delicate spatiotemporal regulation of MMP expression and activity governs diverse processes in normal and pathological conditions such as embryonic development, wound repair, CNS injury, cancer, epilepsy and so on. 63 Emerging evidence suggests that perineuronal nets (PNNs) are dynamically formed and degraded and show periodic condensation and fading, for example, with cycling maternal hormones 64 and during the circadian rhythm. 65 Proteases appear to be pivotal for the physiological PNN malleability. In pathological scenarios, several studies show that epileptiform activities are associated with increased MMP activity and consequently PNN disruption.8,37,38,39,40,62,66,67 Inhibiting MMP activity using wide-spectrum blockers such as doxycycline,11,37 minocycline, 37 and GM6001, 8 or more specific blockers such as IPR-179 62 prevents the PNN degradation and attenuates seizures or neuronal hyperactivity. Furthermore, MMP9 deficiency retards, while overexpression increases seizure propensity. 39 Deficiency of other proteases such as tissue plasminogen activator (tPA) reduces seizure progression and mossy fiber sprouting. 68 These studies provide compelling evidence for MMPs being a master regulator of PNN dynamics; however, mechanisms whereby seizure or epileptogenic events trigger MMP upregulation 40 need further study.
Taken together, these studies provide overwhelming support for the notion that ECM remodeling and PNN disruption is an integral part of epileptogenic changes. Yet mechanistically, how these contribute to seizures, and epileptogenesis still remains to be explored. However, based on the currently available literature, we suggest the following mechanism to work either singly or in combination.
Owing to their negative charges, PNNs have been suggested to act as an “ion filter” that alters the relative mobility of ions in the ECS.14,73 It is easy to envision how a change in K+ and Na+ dynamics would directly affect the firing of PV neurons. 15 Cl ions present another example of how stationary negative charges on ECM can alter the intracellular Cl−concentration via Donnan forces, which is sufficient to increase intracellular Cl− in hippocampal neurons to make GABA less inhibitory and contribute to hyperexcitability. 21 On similar grounds, negatively charged extracellular glutamate (Glu) would be repelled by the PNN’s stationary negative charges to effectively shield PV neurons from Glu excitotoxicity, as evidenced by the fact that CSPGs do not adsorb Glu and prevent neuronal death induced by Glu. 74 Any cleavage of the PNNs consequently would make such neurons more likely to succumb to Glu excitotoxicity, for example, explaining the preferential loss of PV neurons near tumors. 8 Synaptic activity requires pre and often postsynaptic entry of Ca2+, and ECM and PNNs are shown to restrict the Ca2+ diffusion in the ECS. 73 It is possible that temporary binding of Ca2+ to the negative charge of the PNNs would limit the availability of Ca2+ thereby affecting neurotransmitter release.
Parvalbumin neurons are the major inhibitory neurons in the CNS that enforce a powerful inhibition due to their fast-spiking and strategic placement of synapses onto principal neurons. 55 In a recent study on glioma-associated epilepsy, we found that PV neurons that were stripped of their PNNs by MMP release from gliomas have an increased membrane capacitance. 8 This was explained by the PNNs acting as electrostatic insulators, thus changing the dielectric constant of the membrane. Akin to myelin on the axon, this resulted in a decreased membrane capacitance, which also explains how intact PNNs allow cells to fire at almost supraphysiologic frequencies. The degradation by tumors, however, stripped cells of the negative charges, increased capacitance, and slowed their firing thereby tipping the E-I balance toward hyperexcitability (Figure 2). Other conditions such as injury or inflammation may similarly present with release of proteolytic enzymes from glia, microglia, or invading immune cells and may similarly lead to seizures. The PNNs stabilize the excitatory input onto their enclosed GABAergic neurons and any alteration in their structure or components6,26 would allow synaptic contacts to change, and subsequently may facilitate extensive rewiring as suggested for several forms of epilepsy. 6
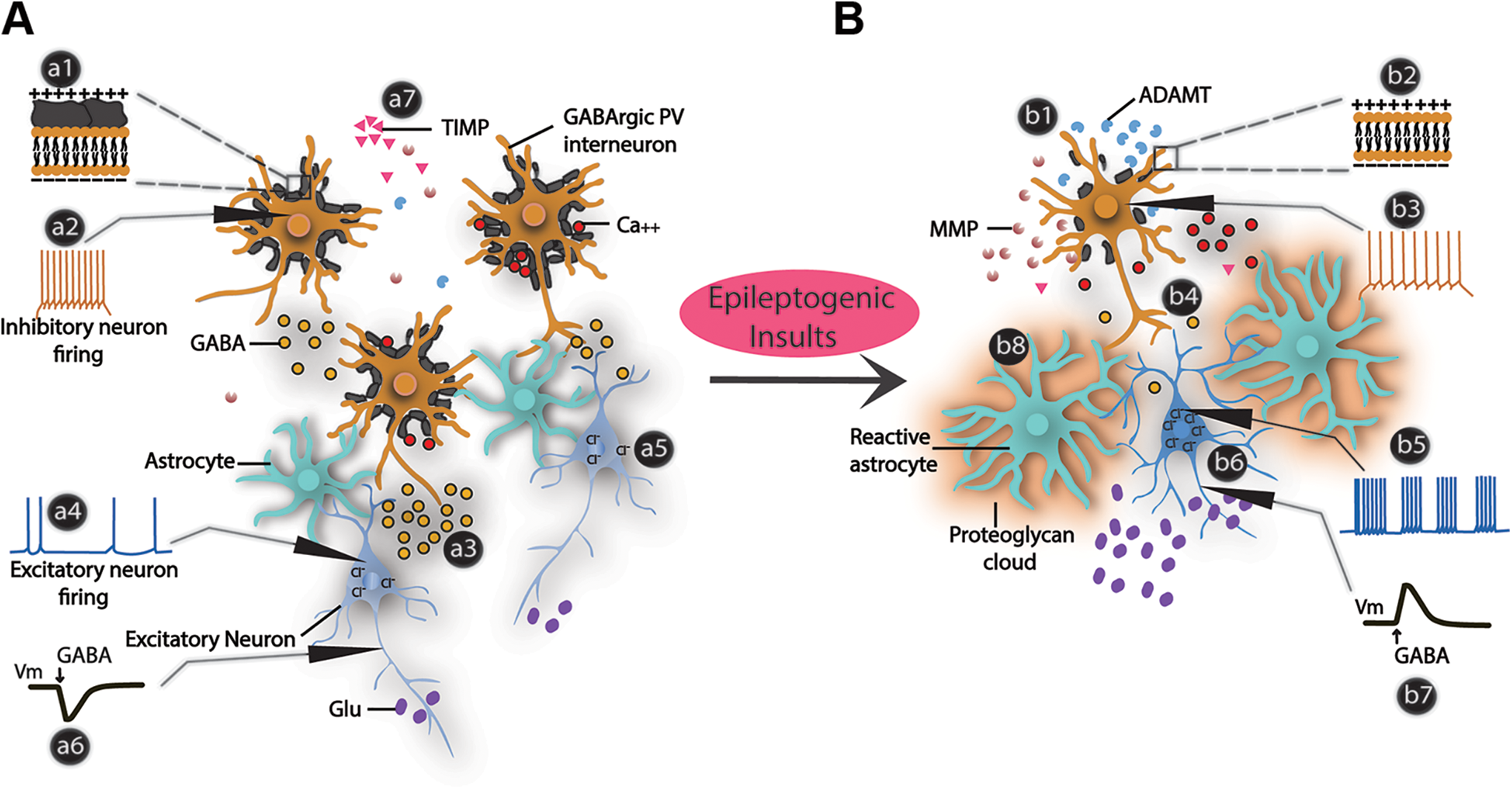
Function of PNNs in physiology and epilepsy. A, In normal physiological conditions, PNNs around the PV interneurons decrease their membrane capacitance (a1), allowing them to generate a high spike frequency (a2) to release sufficient GABA (a3) to balance the excitatory drive (a4). Intact high-density negative charges on ECM and PNNs also maintain a low intracellular Cl− concentration in the principal neurons (a5) retaining a hyperpolarization effect of GABA (a6). A delicate balance of MMPs and TIMPs maintain the normal density and architecture of the PNNs (a7). Intact PNNs also interact with extracellular cations including calcium ions. B, Epileptogenic insults such as traumatic brain injury, glioma, stroke, and so on decrease TIMPs and increase the extracellular MMPs and ADAMTs that cleave the PNNs (b1) and consequently increase capacitance (b2), decrease spiking ability (b3), and reduce GABA release (b4). Prolonged deficiency of GABA release from PV neurons gradually builds-up the excitatory drive (b5) to generate hyperactivity. ECM disruption also alters the Cl− homeostasis in principal neurons (b6) causing GABA to depolarize (b7) them and increase excitatory drive (b5). Reactive astrocytes after epileptogenic insults secrete proteoglycans and extracellular proteases, causing ECM and PNN remodeling (b8). ADAMTs indicate a disintegrin and metalloproteinase with thrombospondin motifs; ECM, extracellular matrix; MMPs, matrix metalloproteinases; PNNs, perineuronal nets; PV, parvalbumin; TIMPs, tissue inhibitors of metalloproteinases.
Summary
Perineuronal nets emerged as an important regulator of neuronal plasticity and PV neuron functions. Several lines of evidence support a causal involvement of PNNs in seizure etiology and suggest PNNs as a potential future therapeutic target.
Highlights
Perineuronal nets are polyanionic sulfated proteoglycan-rich extracellular matrix structures primarily around cortical fast-spiking GABAergic PV interneurons.
Perineuronal nets regulate neuroplasticity and confer neuroprotection, ion-buffering, and aid the PV neuron’s function by regulating its excitability and ion channel expression. Perineuronal nets are dynamic structures and extracellular proteases cleave PNNs in physiology, injury, and disease. Increased protease activity cleaves PNNs in epilepsy, and disruption of PNNs in otherwise normal brains causes hyperexcitability. Preventing PNN disruption by blocking protease activity attenuates seizures and can be explored as a potential therapy.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article: This work was supported by NIH Grants 1R01NS036692-01A1 and 1R01CA227149-01A1.