
Abstract
Voltage-gated sodium channels (VGSCs) are foundational to excitable cell function: Their coordinated passage of sodium ions into the cell is critical for the generation and propagation of action potentials throughout the nervous system. The classical paradigm of action potential physiology states that sodium passes through the membrane only transiently (1-2 milliseconds), before the channels inactivate and cease to conduct sodium ions. However, in reality, a small fraction of the total sodium current (1%-2%) remains at steady state despite prolonged depolarization. While this persistent sodium current (INaP) contributes to normal physiological functioning of neurons, accumulating evidence indicates a particularly pathogenic role for an elevated INaP in epilepsy (reviewed previously 1 ). Due to significant advances over the past decade of epilepsy research concerning the importance of INaP in sodium channelopathies, this review seeks to summarize recent evidence and highlight promising novel anti-seizure medication strategies through preferentially targeting INaP.
Keywords
Voltage-Gated Sodium Channels
Initiation and propagation of action potentials require tightly controlled activation, inactivation, and recovery of VGSCs. 2 Opening of VGSCs is necessary for the depolarizing upstroke of the action potential, whereas channel inactivation, along with potassium channel opening, is required for the repolarizing downstroke of the action potential, allowing recovery of VGSCs from the inactivated state. Proper neuronal function requires that channel properties are fine-tuned to permit cycling through these states: Even small perturbations in VGSC biophysical properties, whether by post-translational modification, pharmacological agents, or by genetic mutations, can lead to profound changes in neuronal excitability.
VGSCs are dimeric transmembrane protein complexes formed of two 260 kDa α-subunits each associated with 1 or 2 auxiliary 30 to 40 kDa β-subunits. 3,4 Each α subunit has 4 homologous domains, each containing 6 transmembrane segments (S1-S6). To date, 10 distinct α-subunit isoforms have been characterized: NaV1.1 through NaV1.9 in addition to the salt-sensing NaX. 5 Of these, NaV1.1, NaV1.2, NaV1.3, and NaV1.6 are highly expressed in the central nervous system (CNS). Considering that each isoform has unique biophysical properties, differential expression of the isoforms, based on cellular population and/or subcellular location, enables significant diversity in neuronal excitability. 2,5
Persistent Sodium Current
A steady-state sodium channel current was first recorded from cat layer V pyramidal neurons and termed the persistent sodium current (INaP). 6,7 Further characterization of INaP revealed properties shared with the transient sodium current such as inhibition by tetrodotoxin and the quaternary derivative of lidocaine, QX-314, as well as unique properties including subthreshold activation and susceptibility to slow, but not fast inactivation. 8
A number of hypotheses have been put forth regarding the molecular mechanism of INaP (originally presented in the study by Crill 9 ): (1) The channel that generates INaP is unique to that which produces the transient sodium current, (2) INaP is produced by the “window current” predicted by the small overlap in the Hodgkin-Huxley activation and inactivation gating variables, and (3) VGSCs stochastically enter an alternative gating mode in which repetitive channel openings generate a “persistent” steady-state sodium current at the population level. Because INaP has been observed in every VGSC isoform (albeit in different relative amounts), and that INaP does not follow mathematical predictions based on the window current, the modal gating hypothesis of the INaP remains the leading hypothesis. Nonetheless, a detailed understanding of the structural basis for INaP is still needed.
INaP critically contributes to the normal repertoire of neuronal excitability. It has been linked with action potential bursting as well as neuronal pacemaking in various neuroanatomical regions. 10 -13 Additionally, INaP contributes to amplification of synaptic currents, both excitatory and inhibitory to enable nonlinear integration of synaptic depolarizations. 14,15 That INaP is important for single neuron neurocomputation relevant for behavior is illustrated by a recent study which demonstrated that INaP is necessary for the amplification of in vivo-recorded spatially-modulated synaptic responses which contribute to place cell activity in CA1 neurons. 16
Regulation of INaP is accomplished by various mechanisms including alternative mRNA splicing, extracellular calcium concentration, protein kinases, G-protein coupled receptors, and oxygen concentration. 17 -23 This wide-array of regulatory factors that influence INaP suggests that its magnitude is dynamically maintained and/or adapted by intrinsic and extrinsic mechanisms. Recent evidence that dendritic INaP increases after tetanic stimulation-induced potentiation supports this notion and implicates INaP as a plasticity product. 24
Persistent Sodium Current and Epilepsy
Since its initial characterization, INaP has been hypothesized to be epileptogenic. One of the early researchers focusing on INaP, Wayne Crill, stated in 1996:
“There is no direct evidence that non-inactivating sodium channels contribute to epileptic behavior. Nonetheless, a direct increase in the non-inactivating component of sodium current would amplify the response of neurons to excitation and could lead to an avalanche of activity in an affected population of neurons” 9
In the decades following this hypothesis, there is now abundant evidence implicating an abnormally large INaP with neuronal hyperexcitability associated with epilepsy (Figure 1). In the following sections, we provide a brief summary of this evidence focusing on voltage-gated sodium channelopathies, as well as pharmacological evidence supporting the rationale behind preferential suppression of INaP to treat epilepsy.
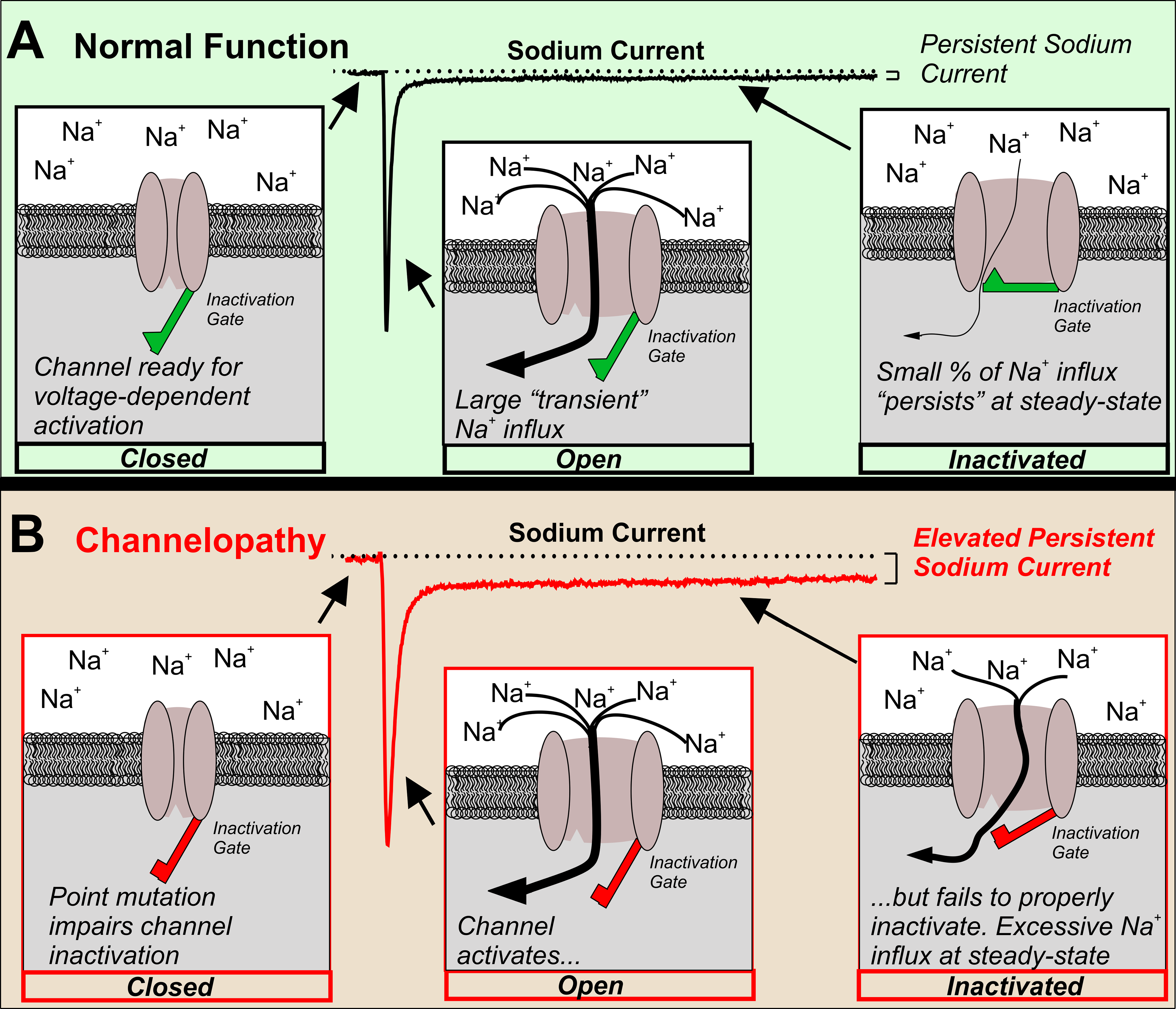
Elevated INaP in an epilepsy-causing channelopathy. In order for neurons to initiate and propagate action potentials, VGSCs must cycle between closed, open, and the inactivated states. (a) Normally, in response to a depolarized voltage step, VGSCs progress from the closed to open states and a large transient INaP is observed. Within a few milliseconds, the inactivation gate closes and channels become inactivated. Note that only a small percentage of the overall INaP remains at steady-state, INaP. (b) An epilepsy-causing channelopathy point mutation impairs inactivation of the VGSC leading to an increased INaP and excessive sodium influx at steady state.
Channelopathies
Because of the critical role VGSCs play in controlling (or driving) neuronal excitability, it is unsurprising that genetic mutations in each of the VGSC isoforms expressed in the CNS has been associated with epilepsy. Biophysical characterization of many of these VGSC mutations has revealed that an abnormal elevation in the INaP magnitude is a common feature of pathogenic ion channels (summarized in Table 1).
Epilepsy-Causing VGSC Mutations Associated With an Increased INaP.

Abbreviations: BFEI, benign focal epilepsy of infancy; BFNIS, benign familial neonatal/infantile seizures; BIS, benign infantile seizures; BNS, benign neonatal seizures; DEE, developmental and epileptic encephalopathy; DS, Dravet syndrome; EE, epileptic encephalopathy; FE, focal epilepsy; GEFS+, generalized epilepsy with febrile seizures plus; OS, Ohtahara syndrome; SMEI, severe myoclonic epilepsy of infancy.
SCN1A
SCN1A, which encodes NaV1.1, is expressed primarily in GABAergic inhibitory interneurons throughout the brain. 52 Mutations in SCN1A are known to cause genetic epilepsy syndromes including generalized epilepsy with febrile seizures plus (GEFS+) as well as Dravet syndrome. 53 Although many epilepsy-causing SCN1A mutations are physiologically loss-of-function (ie, reduced sodium current), numerous SCN1A mutations result in elevations in INaP, and physiologically gain-of-function SCN1A mutations have been recently described to cause an epileptic encephalopathy syndrome more severe than Dravet syndrome. 25 -34 Indeed, an NaV1.1-mediated elevated INaP may contribute to usage-dependent exhaustion of inhibition by facilitating interneuron entry into depolarization block. 25,54
SCN2A
SCN2A, which encodes NaV1.2, is expressed in various neuronal populations and is thought to contribute primarily to backpropagation of action potentials from the axon initial segment to the soma. 55 While both physiologically loss-of-function and gain-of-function epilepsy-causing mutations in SCN2A have been reported, gain-of-function mutations lead to more severe epilepsy syndromes. 56,35 The steady-state INaP was elevated in a number of these variants with or without accompanying shifts in curves for activation or fast-inactivation. 35 -41
Strong evidence that an aberrant INaP is able to cause epilepsy per se was provided by Kearney and colleagues in which a transgenic mouse was generated harboring an engineered mutation (GAL879-881QQQ) known to cause an elevation in INaP. 57 These mice exhibited focal seizure activity in the hippocampus and premature lethality relative to mice expressing wild-type VGSCs. Taken together with studies involving patient-derived mutation characterization, a significant elevation in NaV1.2-mediated INaP is likely sufficient to cause spontaneous seizures and lethality.
SCN3A
SCN3A encodes, NaV1.3, the prominent sodium channel isoform expressed during early development. 58 For this reason, the mild phenotype of NaV1.3 knockout mice has been considered surprising. 59 However, electrophysiological characterization of a number of patient-derived SCN3A variants has demonstrated augmentations of INaP. 42 -45 Interestingly, it was found that treatment of cells expressing mutant SCN3A variants with VGSC blockers lacosamide and phenytoin was able to reduce the aberrantly large INaP, further supporting the causative role for this current underlying SCN3A epilepsy. 42 More detailed insight into SCN3A-related epilepsy will be facilitated by the generation and characterization of transgenic mouse models harboring patient-derived mutations.
SCN8A
SCN8A encodes NaV1.6, the channel most traditionally associated with INaP. 60 The first SCN8A epileptic encephalopathy patient was reported in 2012 with the point mutation N1768D. When expressed in cell lines, the mutation was observed to generate an INaP at substantially larger magnitudes than that observed in the wild-type channel. 46 In the subsequent years, over 300 SCN8A encephalopathy patients have now been identified and numerous SCN8A variants have been characterized and shown to exhibit an INaP with an abnormally large magnitude. 47 -51
Two mouse models harboring patient-derived SCN8A variants have been generated by the Meisler Lab, the Scn8aN1768D (D/+) mouse, which globally expresses the variant mutation, and the Scn8acondR1872W (W/+) mouse, which conditionally expresses the R1872W mutation under the control of Cre recombinase to achieve cell-type or region-specific expression of the mutation. 61,62 Both mouse models exhibit many of the pathological phenotypes seen in patients, including spontaneous seizures and sudden death. Characterization of VGSC function and neuronal physiology from these mouse models has indicated that the abnormal excitability (abnormally large action potential bursts and spikelets) is observed alongside significant elevations in INaP. 60,62 -65
Acquired Channelopathies
The presence of an abnormally large INaP has also been observed in models of acquired epilepsy induced in genetically normal rodents. Induction of status epilepticus leads to an increase in INaP in hippocampal CA1, medial entorhinal cortex, layer V cortex, and subiculum neurons. 66 -70 Fascinatingly, elevations in INaP were observed after the induction of status epilepticus but before the occurrence of spontaneous seizures, supporting the notion that an elevated INaP is causally implicated in epileptogenesis. 67,70
Studies using resected human neurosurgical tissue from a patient with temporal lobe epilepsy have supported rodent experiments, in which recordings from subiculum neurons had a large magnitude INaP that was ∼50% of the transient sodium current magnitude in a subset of neurons. 71 Such an extraordinarily large INaP (relative to neurons from rodent models) observed in human epileptic tissue is further supportive of a role for INaP in human seizure pathology. Gaining a deeper understanding of how INaP participates in human neuronal physiology will provide much-needed insight into the mechanisms by which alterations in INaP contribute to epileptogenesis and epilepsy.
Inhibition of the Persistent Sodium Current to Treat Epilepsy
Beyond evidence from various channelopathies, the importance of INaP in epilepsy is supported by translational studies in which various anti-seizure medications (ASMs) that are effective at suppressing seizures, also inhibit INaP. Perhaps more surprising is that some ASMs that do not act primarily via VGSC inhibition have been shown to modulate INaP, perhaps through downstream regulation. 72 -76 ASMs known to inhibit INaP are summarized in Table 2.
Anti-Seizure Medications that Inhibit INaP.
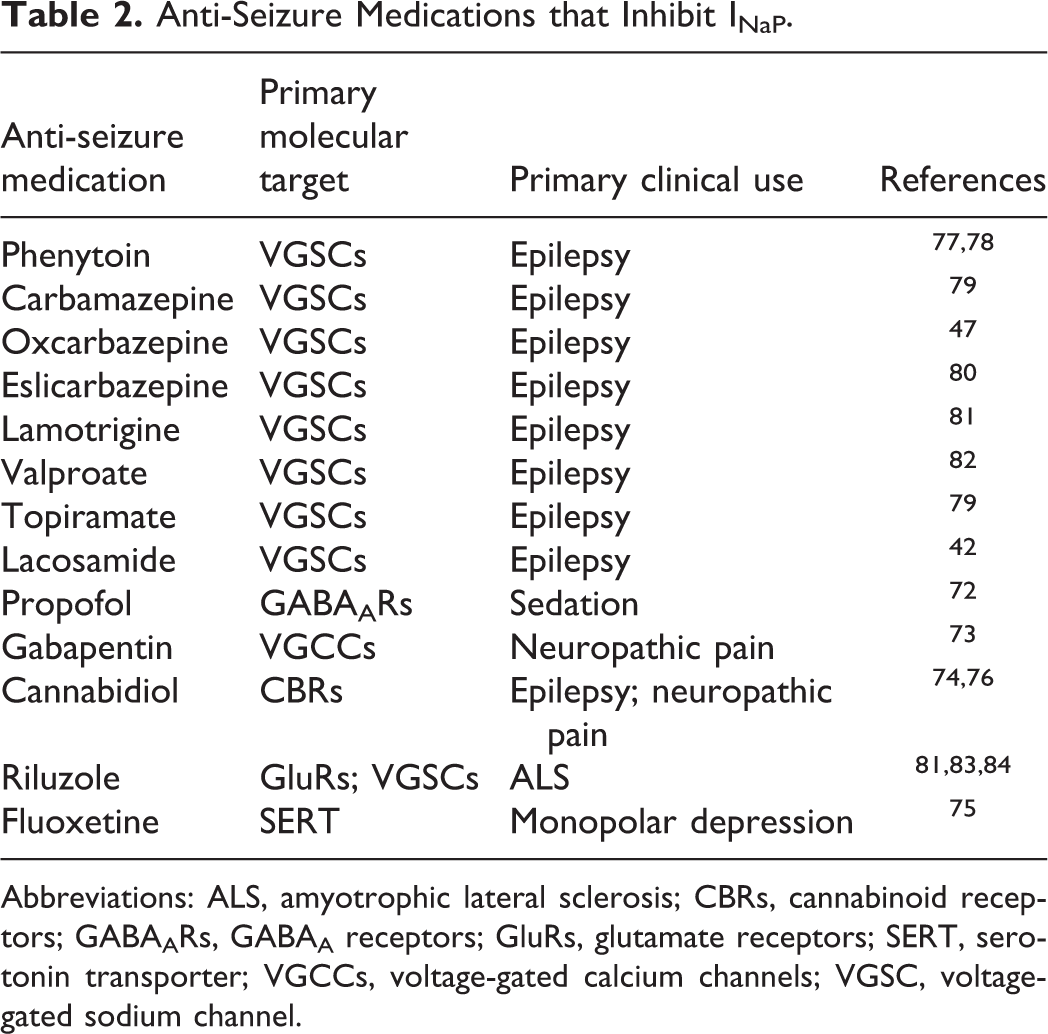
Abbreviations: ALS, amyotrophic lateral sclerosis; CBRs, cannabinoid receptors; GABAARs, GABAA receptors; GluRs, glutamate receptors; SERT, serotonin transporter; VGCCs, voltage-gated calcium channels; VGSC, voltage-gated sodium channel.
A technical barrier to gaining more detailed experimental insight into INaP has been the lack of pharmacological inhibitors that selectively inhibit INaP with little effect on the transient sodium current. To this end, numerous studies have utilized riluzole, an approved amyotrophic lateral sclerosis treatment drug shown to inhibit INaP. 83 -85 However, because riluzole blocks glutamatergic neurotransmission interpretation of some of these studies is complicated. 86
More recently, significant strides toward developing a pharmacological inhibitor that preferentially reduces the persistent component of the total sodium current have been made. GS967, now known as Prax330, was initially developed to preferentially target INaP in the cardiac sodium channel isoform NaV1.5 to treat cardiac arrhythmia syndromes. 87,88 More recently, however, Prax330 has been shown to inhibit INaP in the CNS VGSC isoforms. 65,88 -90 The precise molecular mechanism of Prax330 remains somewhat unclear: An engineered mutation that prevented binding of Prax330 to the local anesthetic site, the location typically associated with sodium channel blockers, failed to prevent its inhibition of INaP despite disrupting Prax330’s use-dependent inhibition of the total sodium current. 91
Prax330 is efficacious in suppressing seizures in mouse models of epileptic encephalopathy harboring a number of VGSC variants including Scn8aN1768D , Scn8acondR1872 W , Scn1a +/-, and Scn2aQ54 . 62,64,92,93 At the level of neuronal physiology, Prax330 has been shown to suppress spontaneous excitability, normalize abnormal action potential waveforms in hippocampal neurons, depolarize action potential threshold, and inhibit synaptic excitability in mutant neurons. 65,92,93 Not only is Prax330 a powerful novel ASM, its ability to preferentially inhibit the INaP renders it a powerful tool to elucidate the importance of INaP on normal neuronal physiology. 94
Future Directions
INaP has been intensely investigated in the past 40 years leading to significant experimental, technological, and pharmacological advancements to more fully understand its role in the nervous system. Nonetheless, important questions remain. For one, structural evidence for modal gating of VGSCs is still needed to further confirm this model of INaP generation. Progress in the structural biology of VGSCs alongside a nuanced understanding of how various channelopathy variants alter biophysical properties will improve our ability to predict neuronal pathophysiology given a particular epilepsy-causing channel mutation.
Another intriguing unknown is the molecular mechanism for pharmacological inhibition of the INaP. Considering that binding to the local anesthetic site was not required for inhibition of the INaP by Prax330/GS967, 91 the binding site(s) responsible for this pharmacological effect remains to be identified and fully characterized.
An additional direction for the near future is to characterize the role of INaP in specific neuronal populations and how these cell types might contribute to seizures and epilepsy. The vast majority of research on INaP has focused on pyramidal neurons from the cortex and hippocampus, yet other cell populations, including inhibitory interneurons, experience profound effects of INaP on their excitability. 13 Taking account of the impact of an elevated INaP on various cell types will be particularly relevant in the epilepsy context in which hypersynchronous neural circuits are generated by interactions between diverse neuronal subpopulations.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors declare the following potential conflict of interest with respect to the research, authorship, and/or publication of this article. M.K.P. has received prior research funding from Praxis Precision Medicines
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institutes of Health Grants R01NS103090 (MKP) and 1F31NS115451-01 (ERW).