
Abstract
Microglia are the resident immune cells and professional phagocytes of the central nervous system. However, little is known about the contribution of their phagocytic signaling to the neuropathology and pathophysiology of epilepsy. Here, we summarize and discuss the implications of recent evidence supporting that aberrant microglia phagocytic activity and alterations in phagocytosis signaling molecules occur in association with microglia–neuronal contacts, neuronal/synaptic loss, and spontaneous recurrent seizures in human and preclinical models of epilepsy. This body of evidence provides strong support that the microglial contribution to epileptogenic networks goes beyond inflammation, and suggests that phagocytic signaling molecules may be novel therapeutic targets for epilepsy.
Introduction
Microglia are the macrophages and professional phagocytes of the central nervous system (CNS). Under physiological conditions, microglia are typically highly ramified cells with dynamic processes that actively monitor their local environment to safeguard neural homeostasis. 1 Under pathological conditions, including seizures and epilepsy, microglia become reactive, develop amoeboid shapes, and produce inflammatory mediators such as cytokines, chemokines, and complement proteins. 2 Depending on their intensity and duration, these inflammatory signals can have beneficial or detrimental effects on the plasticity and survival of nearby cells. 3 For example, short-lasting inflammation can promote neuroprotection by attracting microglia to remove (phagocytose) dead/apoptotic cells, a process that suppresses production of pro-inflammatory cytokines, stimulates release of anti-inflammatory mediators, and promotes tissue repair. 3,4 In contrast, exacerbated long-lasting inflammation is linked to pathological consequences including neurodegeneration, cognitive decline, seizures, and epilepsy. 2,3 Interestingly, new findings support that in addition to inflammatory molecules, signals regulating microglial phagocytic and proliferating properties are altered in response to seizures and may play important roles in epileptogenic processes. Here, we summarize and discuss the implications of these new discoveries.
Phagocytic Signaling
Phagocytosis is the process in which phagocytes, such as microglia, engulf and remove unwanted particles and dead cells. Phagocytosis can be performed by ramified and amoeboid “reactive” microglia, and is orchestrated by an assortment of molecules which regulate chemoattraction, engulfing, and degradation, also known as “find-me,” “eat-me,” and “digest-me” signals, each recognized by specialized receptors (Figure 1). 4,5 “Find-me” signals such as nucleotides (e.g., ATP) are sensed by purinergic receptors (P2Y12) and guide microglia to the location of altered neuronal homeostasis. “Eat-me” signals include phosphatidylserine (PS), which is typically externalized to the outer leaflet of the plasma membrane in cells undergoing apoptosis; Protein S (ProS), an opsonin that binds to PS; and complements C1q and C3b. The receptor Mer Tyrosine Kinase (MerTK) recognizes ProS, while complement receptors 1 and 3 (CR1, CR3) recognize C1q and C3b, respectively. These receptors along with the triggering receptor expressed in myeloid cells 2 (Trem2) aid in engulfment and phagocytosis through remodeling the actin cytoskeleton. 4,5 An additional set of signals referred to as “don’t-eat-me” signals include the integrin associated protein CD47 and its receptor the signal regulatory protein α (SIRP-α). It is well-known that phagocytosis of apoptotic cells is anti-inflammatory and contributes to the resolution of inflammation in injured tissues. 4 However, molecules such as C1q, C3b, CR3, and Trem2 can crosstalk with other receptors/pathways to also regulate microglial inflammatory responses, 4 -7 suggesting that depending on the target and context (healthy vs injured) these signals can mediate production of pro- or anti-inflammatory cytokines. Interestingly, a number of studies support that microglial phagocytic signaling is essential for the establishment and maturation of neural networks. 1,7 Importantly, new evidence indicates that dysregulation of these signaling cascades is associated with the pathology of neurodegenerative disorders 1,7 and epilepsy. 8 Recent histological and transcriptomic immune profiling of microglia from patients with drug-resistant seizures showed that microglia have high expression of CR3, Trem2, and MerTK 9 -12 suggesting a robust phagocytic phenotype. In human focal cortical dysplasia (FCD), we found increases in C1q, C3b, and MerTK that paralleled decreases in ProS and Trem2. 13 In addition, decreased levels of CD47 and SIRP-α were found in human FCD and tuberous sclerosis complex (TSC). 14 Taken together these findings suggest that microglia may have altered phagocytic functions in the human epileptic brain.
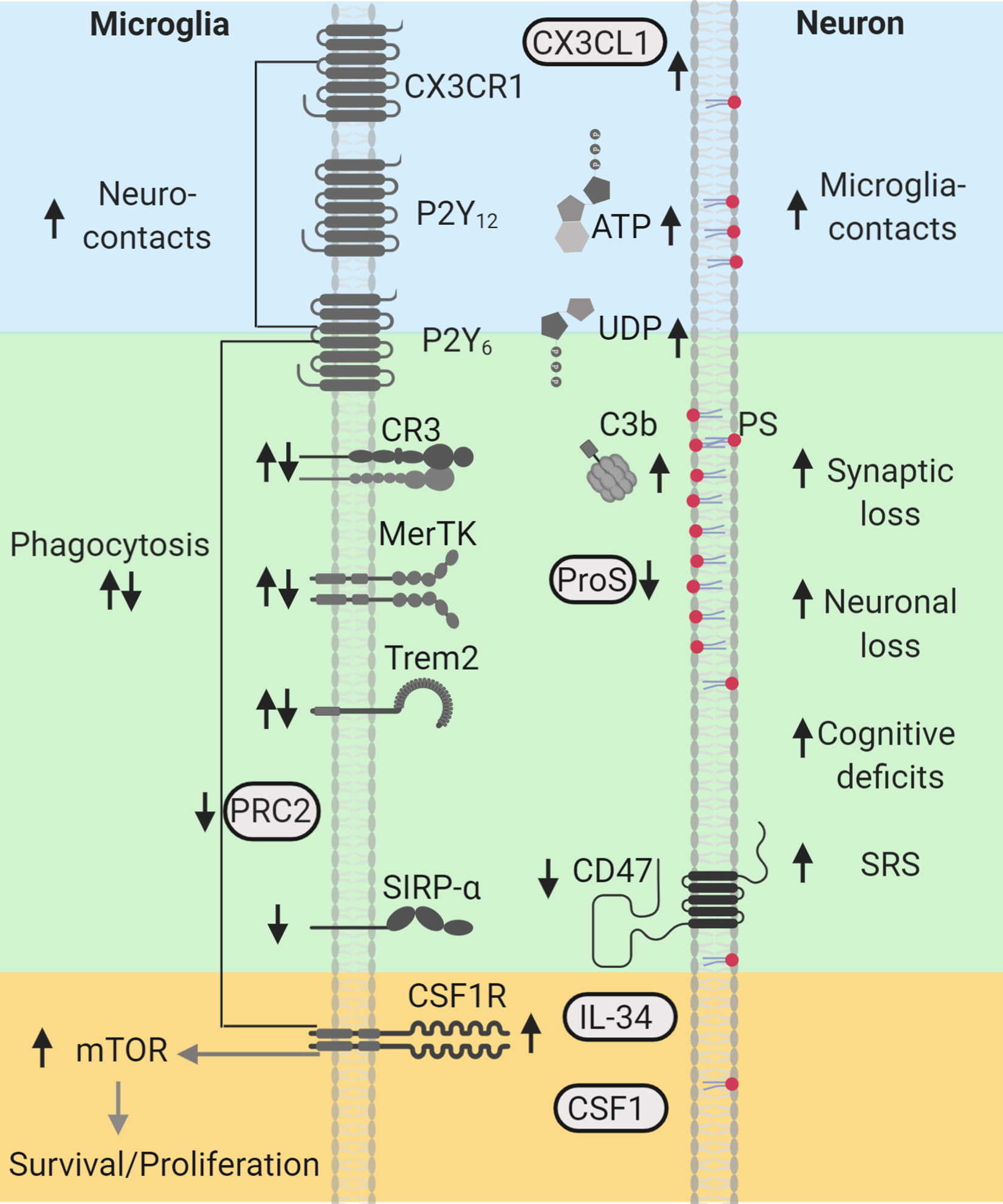
Phagocytic signaling molecules altered in human and experimental epilepsy. “Find-me” signals CX3CL1/CX3CR1, ATP/P2Y12, and UDP/P2Y6, shown in blue, are associated with increased neuroimmune interactions during seizures. Microglia clearance/phagocytic activity controlled by PRC2 and mediated by “eat-me” signals PS (red), C3b/CR3, ProS/MerTK, and Trem2, shown in green, are associated with neuronal/synapse loss, cognitive deficits, and spontaneous recurrent seizures (SRS). “Don’t-eat-me” signals, CD47 and SIRP-α, shown in green, are reduced in human epilepsy. CSF1R-mTOR signaling activated by CSF1/interleuklin-34 (IL34), shown in yellow, regulate microglial survival, proliferation, and phagocytic microglial properties, and are associated with synaptic loss, cognitive decline, and SRS. Arrows indicate the direction of the changes reported in human and experimental models. This diagram was created with Biorender.com. CR indicates complement receptor; CSF1R, colony stimulating factor 1 receptor; MerTK, Mer Tyrosine Kinase; mTOR, mechanistic target of rapamycin; P2Y12, purinergic receptors; ProS, Protein S; PRC2, Polycomb repressive complex 2; PS, phosphatidylserine; SIRP-α, signal regulatory protein α; SRS, spontaneous recurrent seizures; Trem2, triggering receptor expressed in myeloid cells 2.
Activity-Dependent Microglia–Neuronal Interactions
Microglia sense neuronal activity through the purinergic receptor P2Y12R, which responds to microgradients of ATP. 15 Under normal conditions, active neurons release ATP. However, following seizures ATP release increases. 15 Consequently, microglial–neuronal interactions occur in an activity-dependent manner 16 -18 and have been observed in epilepsy. In human epilepsy, microglial processes were found in close proximity and apposed along cortical apical dendrites. 13,19 In experimental models, pronounced microglial interactions with hippocampal CA1 dendrites were observed during and after status epilepticus (SE). 17,20,21 Studies using 2-photon live imaging of in vivo and ex vivo systems demonstrated that microglial processes move rapidly toward dendrites in response to seizures, a process that is dependent upon neuronal NMDA receptor activation along with microglial P2Y12R and fractalkine (CX3CL1-CX3CR1; “find-me” signal) signaling. 17,22 These studies also reported that mice lacking either P2Y12R or CX3CR1 exhibited higher seizure severity during SE, 17,22,23 thereby suggesting a potential role for microglial–dendritic interactions in controlling neuronal excitability. Although the functional impact of these contacts in epilepsy is not definitively known, emerging evidence suggests that microglial contacts can alter the structure of synaptic sites. Live and electron microscope imaging of microglia in cortex and hippocampus demonstrated that direct interactions with synaptic structures can result in the disappearance or growth of pre and postsynaptic elements. 18,24 -26 For example, some microglia contacts were followed by the loss of spines, axonal boutons, or spine head tips (by trogocytosis), 18,24 -26 while others stimulated filopodia growth on dendrites 26 and spine heads. 24 Taken together, these findings suggest that activity-dependent increases in physical microglia–dendritic/synaptic interactions may contribute to circuit remodeling in epilepsy. Thus, determining the functional impact of these neuroimmune interactions may lead to novel treatment strategies to control neuronal hyperexcitability.
Phagocytosis of Synapses
During synaptogenesis in the healthy brain, microglia eliminate extranumerary synapses in an activity-dependent manner to allow for stronger synapses to form functional connections. 7,18,27 -29 Microglial synaptic pruning in developing networks is mediated by “eat-me” signals C1q and C3b 27,28,30,31 and by receptors CR3, 27,28 CX3CR1, 29 and Trem2. 32 CX3CR1, Trem2, C3, or C1q knockout (KO) mice display either reduced microglial engulfment of synaptic material or higher densities of spines/synapses and excessive innervation in cortical or hippocampal networks. 27 -29,32 Functional implications of failed microglial synaptic pruning during development include altered synaptic plasticity, neuronal hyperexcitability, and seizures. 7,30,31 For instance, insufficient synaptic pruning in C1q KO mice is associated with increases in spine density, synaptic connectivity, excitability in the somatosensory cortex, and absence seizures. 30,31 However, higher levels of phagocytic signaling molecules are also associated with synaptic dysfunction. For example, increases in complement C1q and C3/CR3 in mature/adult systems are linked to exacerbated synaptic pruning and cognitive decline, including learning and memory impairments, in models of neurodegenerative disorders. 7 These findings are relevant because increased levels of C1q and C3 are consistently found in human and experimental models of epilepsy, thereby suggesting a potential role for complement proteins in activity-dependent microglial synaptic pruning in seizure disorders. Elevated levels of C1q and C3 occur in human epilepsies with drug-resistant seizures including temporal lobe epilepsy (TLE), 9,33 FCD, 13 and TSC. 34 Similarly, long-lasting increases in the levels of C1q-C3 that correlate with seizure severity occur in adult rodent models of SE and acquired TLE. 33,35,36 It is possible that a complement-dependent microglial elimination of synapses may contribute to the exacerbated synaptic loss, seizures, and memory impairments that occur after SE and in epilepsy. 35 The overall functional impact of aberrant C1q-C3 signaling to epileptic networks would depend on the proportion of excitatory or inhibitory cells/synapses being phagocytosed—an idea that requires further investigation.
Phagocytosis of Neurons
During neurogenesis in the healthy developing brain, a high number of cells undergo apoptosis as neuronal networks develop and mature, and microglia are tasked with their removal. Clearance of apoptotic newborn cells continues in the mature hippocampus, where neurogenesis persists through adulthood. 37 In the adult hippocampal subventricular zone, noninflammatory microglia help maintain homeostasis by rapidly removing excess newborn cells through apoptosis-coupled phagocytosis. 37 However, following SE-induced epilepsy, microglia failed to remove newborn apoptotic cells allowing their accumulation throughout the dentate gyrus (DG). 16 Using 2-photon microscopy along with quantitative reverse transcription-polymerase chain reaction of microglia, Abiega et al showed that this phagocytic impairment paralleled decreased motility, reduced levels of Trem2, MerTK, and CR3, and increased levels of inflammatory cytokines. 16 Despite the reduced phagocytosis of apoptotic cells, microglial engulfment of nonapoptotic viable cells was observed in the DG during epileptogenesis. 16,38 In fact, inflammatory microglia can engulf and “kill” stressed but otherwise healthy neurons in proximity to injured/apoptotic cells. 5 Phagoptosis of viable cells occurs through the transient exposure of PS, 5 which could take place in association with seizures and exacerbate cell loss in epilepsy. Although it is not known whether this contributes to epilepsy, evidence that PS supplements are linked to reduced seizure frequency in aged rats 39 and epileptic patients 40 suggest a potential role. Nevertheless, in contrast to Abiega’s findings, Koizumi et al reported that kainate-induced seizure activity provoked an increase in UDP/P2Y6–dependent microglial phagocytic activity in the hippocampal CA3 region. 41 Because microglial immune properties including clearance/phagocytic activity are region- and context-specific, 8,42 it is possible that these differences may be due to a higher load of dead/dying cells in the CA3 as opposed to DG, where neurogenesis occurs. Lastly, a recent study showed that disinhibition of microglial clearance activity due to ablation of the Polycomb repressive complex 2 resulted in decreased spine density in cortical neurons and spontaneous recurrent seizures (SRS) in aged mice, 42 thereby suggesting that enhanced phagocytic activity may be epileptogenic. Further research is needed to determine how seizures alter the phagocytic profiles of microglia in different brain areas and how these relate to regional neuropathology.
Microglial Proliferation
Microgliosis is the proliferation and accumulation of reactive microglia. Microgliosis is widely observed in human epilepsy as well as in experimental models. 8 To determine how microgliosis contributes to epileptogenesis and seizure generation, it is necessary to examine the regulatory signaling pathways. Activation of the colony stimulating factor 1 receptor (CSF1R) signaling pathway in microglia, by CSF1 or interleukin-34, leads to the downstream activation of a number of molecules including the mechanistic target of rapamycin (mTOR) to regulate survival, proliferation, and phagocytic properties. 6,43 Because mTOR is ubiquitously expressed in neurons and microglia, recent studies have specifically targeted mTOR hyperactivation in microglia 44 as well as CSF1R signaling. 45 -47 A microglia-specific TSC1 KO mouse model produced an mTOR hyperactivation phenotype that resulted in an increase in the number of microglia with enhanced phagocytic activity in the hippocampus. 44 In these mice, the altered microglial properties correlated with reduced densities of excitatory and inhibitory synapses, and with the development of SRS. 44 In a rat model of acquired TLE, we found that at the peak of SE-induced hippocampal microgliosis these cells had activated mTOR signaling and were localized to areas with severe spine/dendritic loss. 21,48,49 Treatment with the mTOR inhibitor rapamycin attenuated the SE-induced microgliosis, dendritic/spine loss, and memory deficits, 21 suggesting that microgliosis contributes to the epilepsy dendritic and cognitive pathology. Neuroprotective and antiepileptic outcomes were also reported when inhibiting CSF1R signaling in mouse models of SE. 45,46 Blocking CSF1R signaling with either CSF1 antibodies or the CSF1R antagonist GW2580 attenuated microglial proliferation and neuronal loss in hippocampi of mice that sustained kainate-induced SE. 46 Similarly, treatment with the CSF1R inhibitor PLX3397 decreased both the number of IBA1-positive microglial cells in the hippocampus and seizure frequency in a mouse model of pilocarpine-induced SE. 45 Although these studies suggest that suppressing microgliosis during epileptogenesis is beneficial, recent studies also showed that total ablation is detrimental, 47,50 thereby suggesting that microglia have multiple roles. Ablation of microglia with the CSF1R inhibitor PLX5622 exacerbated the seizure phenotype and the mortality rate in epileptic mice induced with the Theiler murine encephalomyelitis virus. 47 Moreover, homozygous mutations in the human CSF1R gene are associated with a drastic loss of microglia and brain structural malformations. 50 Of two patients described in this study, one died prematurely at age one and the other presented with developmental delay and epilepsy. 50 Taken together, these data put forward the idea that the wide range of rapamycin-mediated outcomes described in the epilepsy literature may also have do to with the modulation of mTOR-dependent microglial proliferating and phagocytic properties. In addition, these findings suggest that inhibition of microgliosis through the CSF1R pathway during epileptogenesis may be a novel approach to prevent or reduce SRS.
Conclusion
Although microglia are the professional phagocytes of the CNS, relatively little is known regarding their phagocytic profiles in epilepsy or their contribution to the neuropathology and pathophysiology of this disorder. The studies described here are the first to show that seizures promote alterations in phagocytosis-associated signaling molecules that result in neuroimmune interactions, 13,17,19 -22 increases in “find-me” 17,22,41 and “eat-me” 9,13,33 -36 signals, decreases in the phagocytosis of newborn apoptotic cells, 16 phagocytosis of viable newborn cells, 16,38 and increases in microglial phagocytic activity and/or proliferation that parallel neuronal/synaptic loss, seizures, and cognitive decline. 21,35,41,42,44 -46 Thus, it is possible that microglia may modulate synaptic circuitries in epilepsy by improperly phagocytosing synaptic structures and neurons. However, whether this is a pro- or anti-epileptogenic mechanism, or if this is a cause or consequence of seizures and epilepsy requires further investigation. Overall, this body of evidence provides strong support that the microglial contribution to the epileptogenic networks goes beyond inflammation, and suggests that phagocytic signaling molecules may be novel therapeutic targets for epilepsy.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.