
Abstract
The cytokine storm (CS) in hyperinflammation is characterized by high levels of cytokines, extreme activation of innate as well as adaptive immune cells and initiation of apoptosis. High levels of apoptotic cells overwhelm the proper recognition and removal system of these cells. Phosphatidylserine on the apoptotic cell surface, which normally provides a recognition signal for removal, becomes a target for hemostatic proteins and secretory phospholipase A2. The dysregulation of these normal pathways in hemostasis and the inflammasome result in a prothrombotic state, cellular death, and end-organ damage. In this review, we provide the argument that this imbalance in recognition and removal is a common denominator regardless of the inflammatory trigger. The complex reaction of the immune defense system in hyperinflammation leads to self-inflicted damage. This common endpoint may provide additional options to monitor the progression of the inflammatory syndrome, predict severity, and may add to possible treatment strategies.
Keywords
Impact Statement
The current pandemic of COVID-19 or SARS-CoV-2 infection has underscored the fact that the defense system that protects us can be overactivated which leads to an imbalance of protection and self-inflicted damage. The CS that invokes end-organ damage is not unique to COVID-19. Despite different triggers, a common denominator seems to correlate with clinical severity based on extreme apoptosis and ineffective removal of these apoptotic cells. Our defense system in hyperinflammation can be deemed appropriately exaggerated, but can also be pathologically devastating. Data are reviewed of examples of conditions that can lead to hyperinflammation and implicates possible ways to predict severity and may provide tools for the treatment of severely ill patients.
Introduction
Hyperactivation of innate and adaptive immune cells and highly elevated levels of circulating cytokines are often described under the umbrella term cytokine storm (CS) or cytokine release syndrome (CRS). 1 The hyperinflammatory syndrome presents an overlap between inflammatory responses that are deemed appropriately exaggerated and pathologically devastating. 2 The current COVID-19 pandemic has shown the complexity to clearly define the pathology and underlying mechanisms in which hyperinflammatory syndromes lead to end-organ damage via different immune mechanisms. 3 Bacterial or viral infections are well-recognized triggers of inflammation, but also abnormalities in immune response or human physiology can lead to devastating hyperinflammation.4,5 Apoptosis of stressed cells and proper removal is an important factor. While a proper activation of the apoptotic system is important, so is recognition and removal of apoptotic cells. Loss of normal phospholipid asymmetry in apoptosis provides an “eat me” signal to macrophages. While phosphatidylserine (PS) exposure is essential for platelets to induce the hemostatic process, unwanted PS exposure as the result of inadequate removal of apoptotic cells will result in a prothrombotic state, often correlated with severe inflammation-induced illness. Secretory phospholipase A2 (sPLA2), an essential secondary messenger in inflammation, 6 generates the building blocks for inflammatory signaling molecules like eicosanoids and platelet-activating factor (PAF). In addition, sPLA2 will hydrolyze bacterial and viral membranes, and as such assists in host defense. Unfortunately, sPLA2 will also effectively hydrolyze PS exposing (apoptotic) cells. This in turn will release uncontrolled cellular content and lipid breakdown products in the environment with the potential to stress and damage neighboring cells leading to a devastating downward spiral of cellular and organ damage.
Figure 1 shows a working model to illustrate the link between the inflammasome, upregulated sPLA2 levels, induced apoptosis, cellular damage, and the prothrombotic state. The simplified presentation of these processes illustrates the normal reaction on an inflammatory trigger, and defines a common endpoint when the system reacts out of control. The immune system gears up to protect, immune cells respond, cytokines such as Tumor Necrosis Factor alpha (TNFα), Interleukin-6 (IL-6), and other messenger molecules, including C-reactive protein (CRP), is upregulated, sPLA2 provides building blocks for the generation of inflammatory compounds, and assists in the elimination of foreign entities like bacteria or viruses. In hyperinflammation, the normal and continuous process to remove unwanted apoptotic cells can get overwhelmed. The out of balance process to protect leads to a process that initiates a thrombotic state and lead to cellular damage and multiorgan failure.
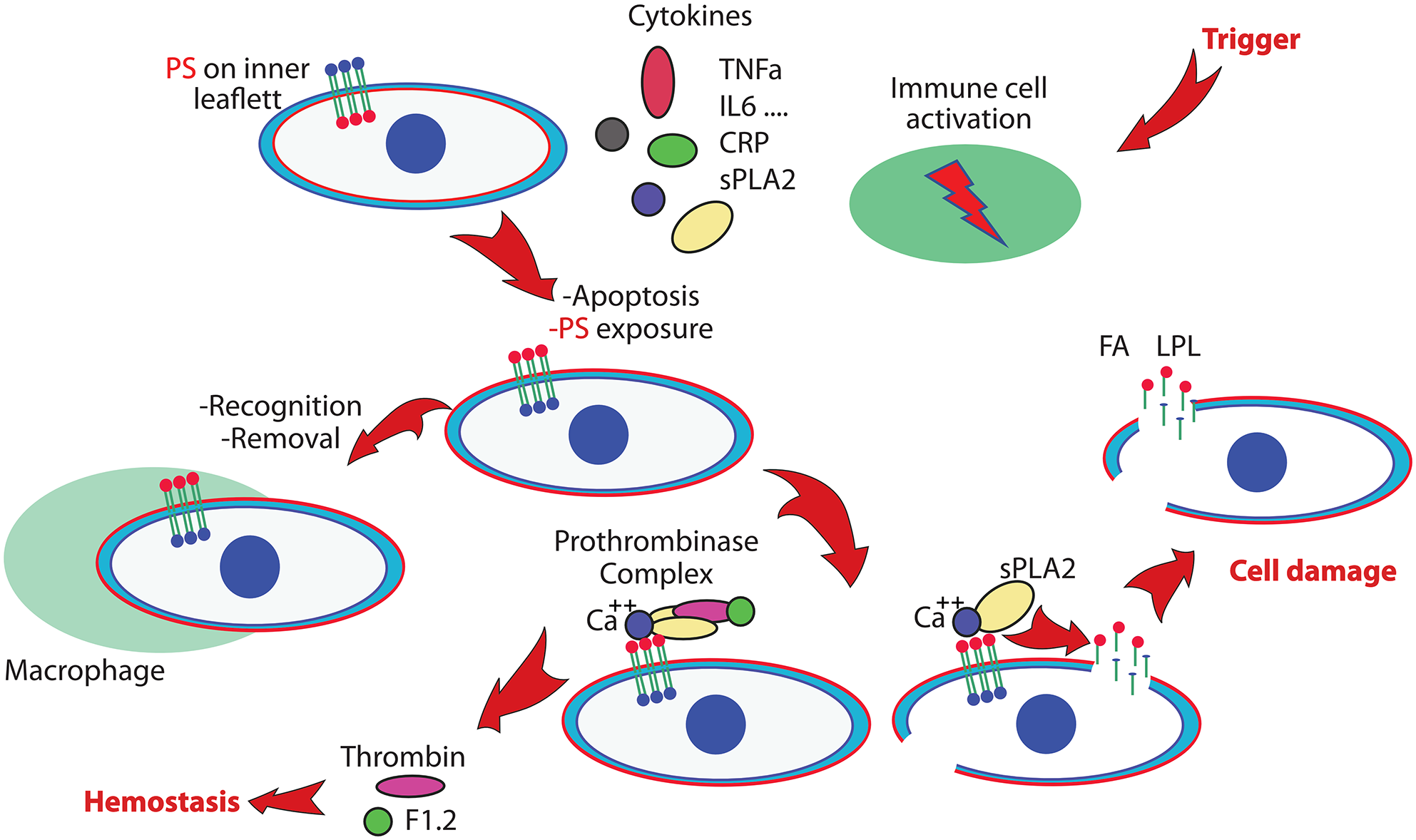
Simplified scheme of processes that will lead to vascular damage and multiorgan failure. An inflammatory trigger activates the immune system and initiates the production of several cytokines and the generation of secretory phospholipase A2 (sPLA2). Apoptotic processes activate phosphatidylserine (PS) exposure, a signal for cell removal. In hyperinflammation, overwhelming numbers of PS exposing cells that are not properly removed will imbalance the hemostatic system by unwanted activation of the prothrombinase complex. PS exposing cells are targets for sPLA2, leading to cell damage and generation of products including Fatty Acid (FA) and Lysophospholipid (LPL) that will damage additional cells. This can lead to a prothrombotic state and multiorgan failure. (A color version of this figure is available in the online journal.)
In this review, we attempt to make the case that despite the various triggers, variants in risk factors, and confusion regarding diagnosis of hyperinflammation pathology, the final step is common and centers on an out of balance system of apoptosis, PS exposure and significantly elevated sPLA2 levels in the inflammasome. This may point at a different way to monitor the clinical state of patients and suggests optional alternative approaches for treatment.
Plasma membrane phospholipids
The plasma membrane phospholipid distribution of animal cells is markedly asymmetric. The amino phospholipids PS and phosphatidylethanolamine (PE) are concentrated in the inner leaflet, whereas phosphatidylcholine (PC) and sphingomyelin (SM) are concentrated in the outer leaflet. Maintenance of membrane lipid asymmetry, and particularly the maintenance of PS in the inner leaflet, is a dynamic process that influences many events over the lifespan of the cell. Flippases or floppases are lipid pumps, that utilize ATP to translocate lipids between the bilayer leaflets. Scramblases, activated when physiologically required, transport lipids in both directions. Together with inhibition of the active inward movement, scrambling results in the loss of lipid asymmetry.7–9
The exposure of PS is an important physiologic event. On platelets, PS stimulates blood coagulation, and PS exposure by other cells during apoptosis provides an “eat me” signal to macrophages leading to “apoptotic clearance.” Programmed cell death is normally an orderly, energy-dependent process that causes cells that have completed their useful functions to die without inducing an inflammatory response. A diverse array of distinct and redundant receptor systems by phagocytes have developed, and provide an efficient recognition and elimination mechanism. The capacity of blood to form thrombin is a critical and a highly regulated determinant in the hemostatic process. PS exposure of platelets leads to plasma thrombin generation, as the prothrombinase complex binds in the presence of calcium to the negatively charged PS exposing membrane surface. The formation of this complex activates the cleavage of prothrombin to thrombin and fragment 1.2. Excessive and unwanted presence of PS exposure will lead to a prothrombotic state, 10 and lack of a proper removal of PS exposing cells can result in uncontrolled and persistent presentation of self-antigens to the immune system. 11
sPLA2 plays an important role in inflammation. The enzyme is a member of the large family of phospholipases that cleave the ester bond on the sn-2 position of phospholipids to generate lysophospholipid (LPL) and free fatty acid (FA). The different members of this family have different capabilities to attack cellular membranes. While the enzymes of snake and bee venom are potent in cellular attack of human membranes, the closely related pancreatic and secretory phospholipase isoforms will not randomly attack human tissue for obvious reasons. 12 However, sPLA2 will effectively hydrolyze bacterial and viral membranes and generate free arachidonic acid as building blocks for a large set of bioactive molecules. 6 sPLA2 levels are highly increased in acute clinical syndromes including sickle cell acute chest syndrome (ACS), pneumonia, acute asthma, and serious bacterial or viral infections.13–21 In contrast to normal cells, PS exposing membranes are rapidly broken down by sPLA2 in a calcium-dependent process. 22 This process can be inhibited by removal of calcium or the presence of proteins that bind in a calcium-mediated fashion to the PS exposing cell surface and “cloak” the PS exposing membrane from the sPLA2 attack or thrombotic activation. 23 Regardless of the trigger, elevated sPLA2 levels indicate a strong ongoing inflammatory signal and suggest a role of this enzyme in cell damage and organ failure.6,13,14,22,24,25 During convalescence, following acute illness, extreme levels of sPLA2 levels return to normal, but may remain elevated in chronic inflammatory conditions like rheumatoid arthritis 26 or sickle cell disease (SCD). 27 In contrast to the level of many cytokines that increases and decreases rapidly, the changes in time of the sPLA2 levels are more moderate. This makes measurement of sPLA2 levels in a good laboratory biomarker for the onset and timeline of clinical severity as was shown in the case of ACS in SCD.14,25,27
Endothelium plays a vital role in vascular physiological processes, but is also a major target to be considered in organ damage. Activation of endothelial cells leads to release of vasoactive substances including nitric oxide, PAF, prostacyclin, mitochondrial N-formyl peptide, and endothelin, as well as mediators of inflammation and thrombosis. Endothelial functions will be altered due to damage of the normal circulation. Ischemia of the endothelial cells and reperfusion due to resuscitation with fluids will induce endothelial cell apoptosis. Deprivation of oxygen required by endothelial cell mitochondria initiates an increase in mitochondrial reactive oxygen species and release of apoptogenic proteins. The structure of endothelium is compromised which, when not repaired, causes an impairment of the protective endothelial barrier resulting in increased permeability and leakage of fluids into the tissue.
Together the exposure of PS and proper recognition and removal of PS exposing apoptotic cells is essential. An imbalance in this highly orchestrated process can lead to a prothrombotic state, uncontrolled breakdown of cells, and generation of products that will lead to a devastating spiral of events that leads to end-organ damage as illustrated in Figure 1.
Hyperinflammation indicates a finely tuned defense system out of control, related to invasion of pathologic and/or deficiencies of the immune response. Sepsis and septic shock are the important causes of morbidity and lethality resulting from the extreme response to an infection. sPLA2 has been implicated as a mediator of organ failure associated with critical illness including intestinal disorders and life-threatening lung disorders such as acute lung injury (ALI) and the Acute Respiratory Distress Syndrome (ARDS).28,29 Since early identification of sepsis is critical, sPLA2 has been proposed as a predictor of sepsis and bacterial infection. 30
Hemophagocytic lymphohistiocytosis (HLH) is a highly fatal condition 31 as the result of mutations in immune cells leading to an abnormal response to inflammatory triggers.32,33 The unregulated activation of macrophages and T lymphocytes culminate in organ damage in part due to the inability to properly remove altered, apoptotic cells. The killing of target cells is a complex, multistage process that concludes with directed secretion of lytic granules-mediating apoptosis. Lack of lytic granules in natural killer (NK) cells is related to serious and often fatal diseases, such as familial hemophagocytic lymphohistiocytosis (FHL) or Griscelli syndrome type 2. 34 The X-linked inhibitor of apoptosis deficiency, XIAP (aka XLP-2), is a rare primary immunodeficiency frequently triggered by the Epstein–Barr virus (EBV) infection. XIAP is an antiapoptotic molecule, and recent findings demonstrate the role of XIAP in innate immunity and in the negative regulation of inflammation.35,36
SCD is marked by vaso-occlusive events as the result of a point mutation in globin which leads to hemoglobin polymerization, membrane changes, increased binding of blood cells to endothelium 37 resulting in an inflammatory and prothrombotic state, and organ damage related to ischemia reperfusion injury. 38 A specific case that illustrates the imbalance of the immune system is ACS in SCD which has parallels with the clinical presentation of ARDS, the devastating consequence of SARS-CoV-2 infection that has cost many lives in the last two years. Pulmonary fat embolism found in patients with ACS 39 results from vaso-occlusion and ischemia reperfusion of bone marrow, delivering an acute increase of apoptotic and necrotic material to the lung microcirculation. The inflammatory response and upregulated sPLA2 creates a situation where generation of LPL and FA increases in the circulation 27 and damages the endothelial and epithelium layer in the lung resulting in a rapidly developing clinical crisis. 40 The levels of sPLA2 and CRP have been shown to be harbingers of this serious condition.14,15,25,41 sPLA2 also correlates with postinjury multipole organ failure and is a marker of infection in febrile children presenting to a pediatric emergency department (ED).13,16
Viruses play a central role in devastating hyperinflammation in humans. Three examples are described in more detail that show the correlation with apoptosis, thrombosis, sPLA2, and end-organ damage. Viruses are opportunistic and unique intracellular pathogens as they fully rely on the host cell for their replication, to complete their lifecycle and potentiate disease. The continuous interactions between the human host and these pathogens during their coevolution have shaped both the immune system and the countermeasures used by pathogens. 42 Viruses hijack host cellular machinery for their replication and survival by targeting crucial cellular physiological pathways, including transcription, translation, immune pathways, and apoptosis. Aspects of three viruses with important public health impacts – Ebola, Dengue, and SARS-CoV-2 – are described. We argue that despite their obviously different modes of infection and pathology, they share a common final pathological feature. All can result in an extreme inflammatory response, unbridled formation of apoptotic cells, which in combination with high levels of sPLA2 and activation of hemostatic factors will play an important role in vascular pathology. Programmed cell death, a key component of the host innate immune response, is an effective strategy for the host cell to curb viral spread. However, many viruses are equipped to take advantage of these pathways in the host to subvert host defense and promote their own propagation. 43 In the past decade, emerging viral outbreaks like SARS-CoV-2, Zika, and Ebola have presented major challenges to the global health system and the current pandemic of COVID-19 has shown an intricate system that involves apoptosis, as well as the side effects of the antiviral strategy by the immune defense system. The choice to describe these three viruses is aimed to make the point of a common thread. Many other viral infections can be considered from a similar perspective. Influenza, commonly known as “the flu,” is a well-known vaccination target characterized by symptoms that range from mild to severe. Influenza infection is often mild and includes fever, runny nose, sore throat, muscle pain, headache, coughing, and fatigue. However, it can also lead to serious complications in people who are at high risk. The raging Spanish flu pandemic that occurred a century ago can be considered as another example how we as humans succumb to our own out of balance defense system.
The CS and immune cell apoptosis results in uncontrolled, systemic inflammation in affected individuals, resulting in apoptosis of cell populations different from immune. 62 Blocking lymphocyte apoptosis is not sufficient to improve survival in EBOV infection, 63 and fatal EBOV hemorrhagic fever results in massive intravascular apoptosis, which develops rapidly following infection and progresses relentlessly until death. 64 The initiation of a CS by the virus along with infection of endothelial cells leads to apoptosis and structural and functional changes that attenuate vascular integrity in many organs including the lungs, heart, liver, and kidney, and the damage of blood vessels of non-human primates at end-stage Ebola shows microvascular lesions and impaired blood supply to the organs further initiating damage. 65 Multiple hemorrhages were formed by diapedesis and fibrin deposition and thrombi were the features of hemostatic impairment, 66 as endothelial damage enhances the coagulation pathway leading to thrombus formation in major vessels and capillaries. 67 Apoptosis indicates that PS in the membrane of the host cell appears to play an important role in the life cycle of EBOV, 68 and lipidome changes were related to clinical outcome. 69 PE synergizes with PS to enhance PS receptor-mediated efferocytosis and virus entry.70,71 Plasma samples from patients infected with EBOV showed modified clotting times over time of infection, implicating PS exposing cells that trigger an unbalanced hemostasis. D-dimer levels elevated during infection with EBOV, returned to normal in patients who survived. 72 The use of thromboelastography showed that two infected patients had developed a marked hypercoagulable state early in their illness, which was treated with low-molecular-weight heparin. 73 Virtually no information is available on a potential role of sPLA2 in the vascular and organ damage observed. However, extreme upregulation of this enzyme can be expected and may confirm that the observed apoptosis, PS exposure, thrombotic events, and end-organ damage confirm the model as indicated in Figure 1.
The virus hijacks host cellular machinery for their replication and survival by targeting crucial cellular pathways,75,78 and the innate immune response to DENV infection induces transient immune aberrant activation, cytokine overproduction, and causes apoptosis and dysfunction of these cells. 79 DENV targets include dendritic reticulum cells, monocytes, lymphocytes, macrophages, hepatocytes, and vascular endothelial cells,80,81 and triggers multiple cell death pathways to produce several inflammatory cytokines that can lead to endothelial dysfunction, and therefore vascular leak. 82 Overproduced IL-6 might play a crucial role in activation of coagulation and fibrinolysis systems, and the enhanced production of sPLA2. DENV seems to efficiently adapt to and manipulate the host systems, and thus controls the survival of the infected cells.83–87 The PDZ domain-binding motif (PBM) in DENV proteins provides a high potential to influence the behavior of the host, based on any of the more than 400 cellular protein isoforms with a protein–protein interaction module (PDZ domain) in the human genome. 88 The DENV non-structural proteins manipulate interactions with several proteins to interfere with antiviral interferon signaling, 74 and inhibit the RNase Dicer. 89 DENV also utilizes various strategies to alter apoptotic pathways including autophagy temporarily sparing the infected cell, providing a suitable environment for the viral replication. 90 Virally induced apoptosis contributes directly to the cytopathogenic effects of DENV in cultured cells. Endothelium-related capillary leakage is triggered by DENV itself or by the host. DENV-induced vasculopathy and the unbalance between coagulation and fibrinolysis activation increase the likelihood of severe hemorrhage in DHF/DSS.77,82,91,92 Endothelial apoptosis may result in hypovolemic shock and downstream apoptosis of endothelium due to deprivation of oxygen required by endothelial cell mitochondria, setting the stage for a series of devastating events, damaging vascular viability in the pathophysiology in DHF and dengue with complication (DCC). 93 The DENV non-structural protein 1 (NS1), causes endothelial leakage, induces P-selectin expression and PS exposure in human platelets by interaction with TLR4, adenosine diphosphate (ADP)-induced platelet aggregation, and enhanced platelet adhesion to endothelial cells and phagocytosis by macrophages. 94 Limited data are available regarding the possible role of sPLA2 in dengue. Secreted phospholipases A2 from Viperid venoms with homology to human sPLA2 show ability to hydrolyze DENV suggesting that human sPLA2 could be involved in viral protection 95 by hydrolyzing the viral lipid bilayer. During the first 120 h of clinical illness, sPLA2 activity associated with PAF levels increases, suggesting a role for sPLA2 and PAF in vascular leak and the pathogenesis of dengue. 21 The coagulation and fibrinolysis systems are activated after DENV infection, and the unbalance between coagulation and fibrinolysis activation increases the likelihood of severe hemorrhage in DHF/DSS. Thrombocytopenia is a major characteristic observed in both mild and severe dengue disease and is significantly correlated with the progression of dengue severity. Together, the unbalanced formation of apoptotic cells, altered hemostasis upregulated sPLA2, and endothelial damage in severe dengue confirm to scheme of events in Figure 1.
COVID-19
A conclusive review to describe the molecular mechanisms that underlie the SARS-CoV-2-induced pandemic can only be incomplete at this time. It is hampered by the complexity of the enormous impact on public health, the arrival of new variants like the Delta variant in the second part of 2021 and the Omicron variant that is currently (January 2022) rapidly taking over as the main viral entity in COVID-19. From a biology perspective, we can only marvel at the effectiveness of this virus to mutate rapidly to a possible less severe but still devastating form, extremely effective in spreading through the human population in its own advantage. The rapid rate of new scientific findings and extreme volume of published new studies makes a conclusive detailed assessment difficult. However, the current studies clearly suggest a common denominator between COVID-19, Ebola and Dengue. Severe COVID is associated with hyperinflammation and a CS. 2 Disease severity and fatality is characterized by an association with thrombosis and vascular damage,3,96–100 the elevation of several cytokines (IP-10, MCP-3, and IL-1-ra CRP), and abnormal liver function. As is the case with the other intruders, COVID-19 intends to circumvent the human host defenses and the severe response leads to self-inflicted damage with devastating consequences that can be related to events depicted in Figure 1. The degree of viremia appears to relate to the clinical severity of COVID-19 and it is logical to assume that the number of apoptotic cells formed is also related to the viral dose. As is the case with Ebola or Dengue, viral infection leads to apoptosis, PS exposure, and is related to the macrophage removal of virus-infected cells.101–105 In a report focused on ACE2 receptors on endothelial cells in COVID-19, 106 apoptosis of endothelial cells and mononuclear cells was clearly apparent. Similarly, as observed for lung damage in SCD patients during ACS, endothelial damage and vascular dysfunction in COVID-19 will affect the vascular health in the lung as well as other organs including the brain. While information on the long-term COVID-19 problems is just emerging, it is tempting to assume that end-organ damage inflicted during the acute phase is the underlying reason for the observed clinical issues in COVID-19 survivors. As suggested under these conditions, when PS exposing cells are not efficiently removed, sPLA2 will break down cells that expose PS. 22 Lipolysis of these damaged cells by sPLA2 will generate non-esterified fatty acids (NEFA) and LPLs and lead to release of cellular content in the environment. These sPLA2-induced cellular breakdown products will affect other cells in the circulation when not properly buffered or removed. A clear correlation was reported between levels of CRP and sPLA2 in patients with SCD that develop ACS. 41 CRP provides a binding site for LPL, and recent data have shown a possible correlation between levels of CRP and severity in subsets of COVID-19 patients.107–109 The correlation between upregulated CRP and severity is, however, not always clear. An extensive formation of LPL and FA will overwhelm the normal buffering of sPLA2 products by CRP and albumin or lipoproteins. sPLA2 levels are correlated to clinical severity in the COVID-19 population, including a pediatric subset.17,19,110–114 Even though they represent only a fraction of the total severe COVID-19 cases, pediatric COVID-19 has become a major public health issue. Diorio et al. 115 demonstrated an elevation in cytokine profiles associated with multisystem inflammatory syndrome in children (MIS-C), in children infected with COVID-19, suggesting that early identification of inflammatory factors like sPLA2 is valuable to monitor disease severity. Recent studies have found a considerable number of hospitalized and critically ill pediatric patients.116–118 This seems even more the case with the current omicron variant. Underlying medical conditions, including immune compromise and cardiorespiratory comorbidities, appear to be at increased risk of pediatric COVID-19. 119 Nevertheless, the association of MIS-C with COVID-19 seems also related to previously healthy children with no underlying comorbidities. 120 MIS-C, a postinfectious hyperinflammatory response to SARS-CoV-2 infection, 121 shows high rates of pediatric intensive care unit (PICU) admission, vasopressor requirements, and mechanical ventilation,122,123 and includes myocarditis, cardiorespiratory failure, and even death. 124 A biomarker of pediatric COVID-19 and MIS-C disease severity to guide patient management would be highly valuable to stratify risk. New evidence is emerging daily that COVID-19 is more than a respiratory disease; it can result in coagulopathy, multiorgan failure and in severely ill patients, and circulatory damage may underlie the still poorly understood long COVID syndrome.
Taken together, the examples of viral-induced inflammation suggest that despite very different molecular and clinical processes, the end result of hyperinflammation appears the inability to remove PS exposing (apoptotic) cells resulting in a prothrombotic state which in combination with sPLA2 can lead to cellular damage, endothelial dysfunction, and end-organ damage
Severity prediction
A clear link between biomarkers and prediction of clinical severity as a diagnostic tool in hyperinflammation has been difficult. A large variety of cytokines and other markers like CRP in addition to white blood cell assessment have been used as biomarkers in inflammation but often fail to link to outcome. Many of the cytokines have short response times. They increase and decrease rapidly making timed sampling complex. sPLA2 has a much slower response and measuring its levels in blood has been used to predict the onset of ACS in SCD. Assessment of apoptosis in vivo has been difficult. PS exposure to identify apoptotic cells in vitro can be readily assessed by the use of fluorescently labeled Annexin V. As a fluorescent derivative annexin is a widely used to visualize apoptotic cells by microscopy or flowcytometry. 125 Radiolabeled Annexin V can be used for in vivo studies in animals. Assessment of PS exposure in blood cells can be accomplished ex vivo, 126 but direct assessment of, for example, endothelium PS exposure in patients is not possible. Indirect assessment of PS exposure can be pursued. Thrombin enters the coagulation cascade after binding of the prothrombinase complex to a PS exposing surface and release of Fragment 1.2 from prothrombin. Downstream biomarkers of thrombotic events like Di-dimer are routinely used as biomarkers, but a more direct approach by measuring F1.2 as the initial step of PS exposure could be contemplated. Similarly, if one assumes that sPLA2-induced damage is a major factor in cellular damage, it seems logical to track the levels of sPLA2 in patients. In SCD, for instance, daily assessment of sPLA2 levels predicted the onset of ACS.14,15,25,27 The assay to measure sPLA2 can easily be introduced as a routine assay in the acute care setting, added to the measurements provided by a clinical lab. The sPLA2 results may avoid delayed diagnosis of hyperinflammatory-related pathology as observed in COVID-19 127 and could provide a tool for the clinician to decide on a course of action to benefit the patient.
Treatment
While viremia obviously triggers the pathology observed in viral infections, clinical data suggest that the response by the immune system plays an important role in the morbidity and mortality of COVID-19. This in turn has generated interest in treatments that mitigate the immune response including corticosteroids and other immunomodulatory agents. However, treatments that act broadly to suppress the immune system have the potential to impede the body’s ability to control the viral infection, and there is no clear consensus as to which inflammatory biomarkers identify appropriate anticytokine treatment. Severely ill COVID-19 patients have been treated with general anti-inflammatory drugs that target various cytokines. However, since cytokines are also essential in eliminating SARS-COV-2, blocking these cytokines pathways could potentially lead to unwanted and poor outcomes. Based on measured CRP levels, 128 anti-inflammatory corticosteroids have only led to improved survival in a subset of patients. Drugs that target specific cytokines or cytokine pathways have been only partly successful. Anakinra which blocks both IL-1α and IL-1β seemed to improve survival.129,130 However, a double-blind study of canakinumab, a monoclonal antibody to IL-1β&0x44; failed to show a improved survival. 131 Whereas Janus kinase inhibition which targets multiple inflammatory cytokines 132 seemed to improve survival, 133 antibodies to either IL-6 or its receptor did not show a clear benefit. 128 Since its discovery in the serum of patients with severe inflammation and in rheumatoid arthritic fluids, sPLA2 as a proinflammatory enzyme has invoked intense interest in selective inhibitors of sPLA2-IIA in the hope of developing new and efficient therapies for inflammatory diseases. 134 Corticosteroids reduce the formation of sPLA2 and thereby may benefit severe cases of COVID-19,135–139 but their use has also been related to adverse effects, 140 as shown in a large cohort of relatively healthy patients from Taiwan. 140 Treatments that address specific inflammatory pathways may be more advantageous.
Since blood levels of sPLA2 correlate with severity, addressing measures to lower elevated levels of sPLA2, together with apoptotic changes in cellular membranes, appears logical. Compounds that affect the formation of sPLA2 including IL-6 blockers such as sirukumab, MEDI5117, sarilumab, ALX-0061, clazakizumab, and olokizumab 141 as well as IL-6 inhibitors such as tocilizumab (TCZ) could be considered. Treatment of a number of autoimmune diseases including ulcerative colitis, rheumatoid arthritis, ankylosing spondylitis, Crohn’s disease, psoriasis, psoriatic arthritis, and Behçet’s disease have included the use of infliximab, a chimeric monoclonal TNF-alpha antibody, by action that has been related to a decrease in the formation of sPLA2. 142 Compounds like Varespladib, an sPLA2 inhibitor, 143 and other specific inhibitors of sPLA2 may warrant further investigation as therapeutic agents.
In 2021, a clinical study for the use of Varespladib in COVID-19 was announced (https://clinicaltrials.gov/ct2/show/NCT04969991). The use of an sPLA2 inhibitor in sepsis demonstrated improved survival in a subgroup of patients who received the drug within 24 h of sepsis-induced organ failure. Results in a larger group with severe organ failure was not significant, 144 confirming that the administration of these compounds is needed before major organ damage has occurred. Diagnostic assessment of sPLA2 levels together with the use of sPLA2 inhibitors may allow a better clinical management.
PS exposure of cells triggers both the sPLA2-induced cellular damage and the prothrombotic state. Therefore, “cloaking” of PS surfaces may represent a strategy to mitigate both dysregulation of hemostasis and sPLA2 attack. Careful management is obviously required. Whereas PS exposure on cells other than platelets may lead to a prothrombotic state, the assembly of the prothrombinase complex on the PS exposing surface of activated platelets is essential for normal hemostasis. The current findings in the COVID-19 pandemic seem to confirm the relationship of apoptosis and hemostatic disorder. Abnormal coagulation parameters in COVID-19 patients from Wuhan 99 were confirmed with additional studies.145,146 Both thrombus formation and elevation of D-dimer was reported in COVID-19. 147 Treatment with low molecular weight heparin appeared to associate with outcomes. 97 Annexin is a common name for a group of cellular proteins that bind to PS exposing membranes in the presence of calcium, and Di-annexin was generated to provide a stronger binding and longer lifetime in the circulation to “cloak” PS exposing surfaces. 23 This compound has proven to be effective in modulation ischemia reperfusion injury in animals,148–153 and has been used in solid organ transplants.150–155 We speculate that this compound could lower both the damage invoked by sPLA2 as well as modulate the onset of thrombotic events.
Ultimately, treatment options for several of these devastating inflammatory conditions will rely on a combination of modalities to avoid the onset of hyperinflammation. This obviously includes measures to avoid infection such as vaccines and other public health measures. Lowering the risk of hyperinflammation includes treatments that inhibit, for example, viral replication, and anticytokine and anti-inflammatory agents. However, for the critically sick patients, the tools in the hands of physicians are limited. One can argue that compounds that specifically target sPLA2 and PS exposing cells may benefit these patients and may also have a positive effect on long-term damage as is observed in patients after the resolve of the acute illness.
In conclusion, measurement of sPLA2 and factors that report on the onset of vascular damage in combination with specific therapeutic compounds targeting sPLA2, as well as those that can modulate vascular damage as the result of the presence of PS exposing cells, warrant consideration to modulate the devastation effects of hyperinflammation.
Footnotes
Authors’ Contributions
F.A.K. wrote and edited the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.