
Abstract
We review the evidence that infectious agents, including those that become dormant within the host, have a major role to play in much of the etiology of rheumatoid arthritis and the inflammation that is its hallmark. This occurs in particular because they can produce cross-reactive (auto-)antigens, as well as potent inflammagens such as lipopolysaccharide that can themselves catalyze further inflammagenesis, including via β-amyloid formation. A series of observables coexist in many chronic, inflammatory diseases as well as rheumatoid arthritis. They include iron dysregulation, hypercoagulability, anomalous morphologies of host erythrocytes, and microparticle formation. Iron dysregulation may be responsible for the periodic regrowth and resuscitation of the dormant bacteria, with concomitant inflammagen production. The present systems biology analysis benefits from the philosophical idea of “coherence,” that reflects the principle that if a series of ostensibly unrelated findings are brought together into a self-consistent narrative, that narrative is thereby strengthened. As such, we provide a coherent and testable narrative for the major involvement of (often dormant) bacteria in rheumatoid arthritis.
Impact statement
Rheumatoid arthritis (RA) is accompanied by long-term inflammation that is mediated by cytokines and cross-reactive (auto-)antigens. Here we suggest one explanation is the presence of a (dormant) microbiome in RA that sheds the highly potent inflammagen, lipopolysaccharide lipopolysaccharides (LPS) to catalyze inflammagenesis, including via β-amyloid formation. We discuss various co-existing features in RA, including iron dysregulation, hypercoagulability, anomalous morphologies of host erythrocytes, and microparticle formation. We review literature and provide coherent evidence that an aberrant blood microbiome in RA has a major involvement in the development, progression, and therefore over-all etiology of the disease.
Keywords
Introduction: Disease background
RA is a complex and heterogeneous disease, sometimes classified as a syndrome with shared clinical manifestations.
1
It is the most common immune-related chronic, inflammatory, autoimmune disease and affects approximately 0.5–1% of the adult population worldwide. It occurs as 20–50 cases per 100,000 annually, most commonly in women over 40.2–5 Although this is not yet apparently a mainstream recognition, a frankly overwhelming amount of epidemiological and experimental evidence, that we shall review here, indicates a microbial origin for RA. The clinical features of RA involve the presence of systemic inflammation, with various imbalances between pro- and anti-inflammatory cytokine activities, which may lead to multisystem immune complications.
4
In RA patients, serum or plasma levels of cytokines are considered to indicate the severity of disease
4
and this pathophysiologic presence of pro-inflammatory cytokines is known to be involved in the degradation of bone and cartilage.
4
Due to the complexity of the disease, treatment and disease tracking after diagnosis is very difficult. In this article, we shall review briefly current knowledge regarding the involvement of cytokines and other markers in RA, which are also the hallmarks of systemic inflammation in all other inflammatory conditions. We also discuss clot hypercoagulability and platelet and erythrocyte (RBC) changes, that is consequent upon this persistent systemic inflammation, and how microparticle formation (from both platelets and RBCs) is a characteristic feature of RA. We then review a considerable literature (summarized in part in two books6,7) that suggests that the presence of a variety of detritus produced from walls and membranes of Gram-negative and other bacteria (including wall-less forms) may play a fundamental role in RA development, as well as the accompanying cardiovascular disease and systemic inflammation seen in RA. We discuss how the exposure of genetically susceptible individuals to environmental factors (1) that can act as triggers (2), cause an immunological reaction, followed by an autoimmune response (3), can result in RA (4). We review a plethora of evidence, collectively referred to as Ebringer’s theory (5), that points to the environmental trigger as microbial (particularly from e.g. urinary tract infections) (6). We then look at the role of LPS from these microbes (7) in causing an imbalance between pro- and anti-inflammatory cytokines, followed by systemic inflammation, and the effect on the cardiovascular and hematological health of the RA patient (8) (see Figure 1). Finally, recognizing the lack of easy and accessible biomarkers, we suggest that in a truly precision medicine approach,
8
hypercoagulability and also microparticle presence, as well as LPS and β-amyloid analysis could play an important role in tracking the progression of the disease. A MEDLINE, Google Scholar, Scopus, and Web of Science search was done to review the literature.
Genetically susceptible individuals exposed to environmental factors (1) that act as triggers (2) cause an immunological reaction followed by an autoimmune response (3) that may lead to rheumatoid arthritis (RA) (4). We discuss Ebringer’s theory (5), suggesting the cause of the trigger being microbes (6) and the role of LPS (7) that may result in an imbalance between pro- and anti-inflammatory cytokines, followed by systemic inflammation and the effect on the cardiovascular and hematological health of the RA patient (8). (A color version of this figure is available in the online journal.)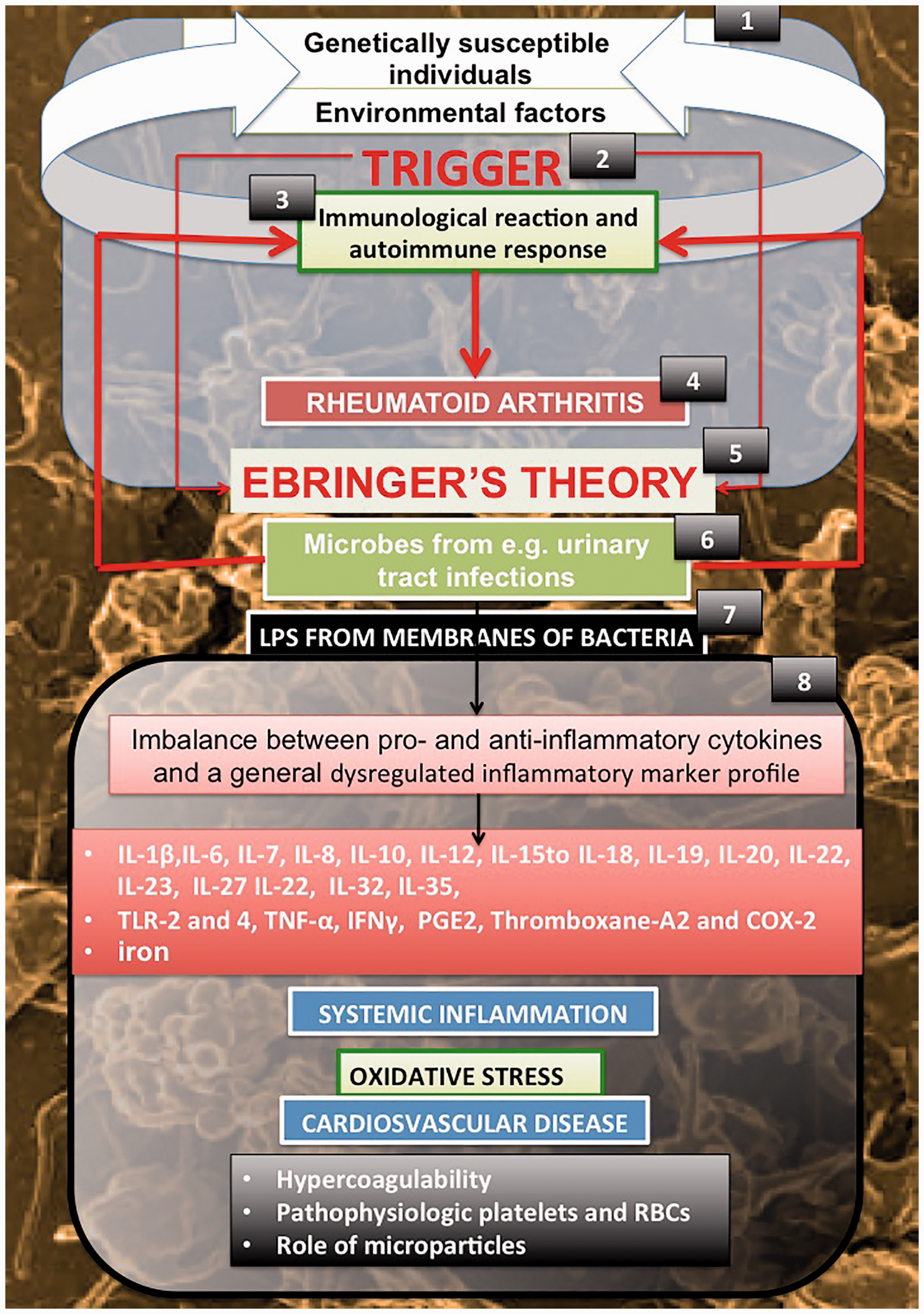
Epidemiology
An initial analysis of the potential causes of a disease is wise to explain any unusual epidemiological features it may possess, 9 following appropriate controls for their veracity. 10 Clearly, within an overall prevalence of ca 0.5–1%, one feature of this disease is its considerable predilection for women (69% of cases in a recent UK survey 5 ) over men, despite some reduction in female prevalence attributed to the use of oral contraceptives.11,12 This is a striking difference of approximately two-or three-fold (see also literature2,12–15). Clearly, it might be linked to hormonal differences, or to something on the X-chromosome, but we know of no persuasive study that suggests what that might be.
Twin studies “show concordance rates of 15% to 30% between monozygotic twins and 5% among dizygotic twins, 14 suggesting that 50% to 60% of RA cases are due to genetic factors.16,17” Other studies comparing monozygotic twins alone show an occurrence in a second twin, when a first twin manifests the disease, as just 12% in Finland, 18 15% for the UK, 19 and 10% in Denmark. 20 Thus, environmental influences have a major influence. Consequently, the leading hypothesis for RA (and indeed for most other autoimmune disorders) is that RA is the result of an environmental exposure or “trigger” in a genetically more susceptible individual, that causes an immunological reaction to the triggering antigen that happens to share one or more epitopes with a host protein (and see Kell and Pretorius 21 ), thus manifesting the autoimmune responses. What might be the most common triggers? One possibility to link the triggers with infection is to look at the presence of flares. Flares are defined as a worsening of signs and symptoms of sufficient intensity and duration to lead to changes in therapy as per the Outcome Measures in Rheumatology Clinical Trials (OMERACT) RA Flare Definition Working Group, developed at OMERACT 9 in 2008. 22 This working Group developed a standardized method for description and measurement of “flare in RA” to guide individual patient treatment. 23
A very high proportion of sufferers were actually exposed to an infection before their disease was diagnosed, but sadly these kinds of data are not typically recorded properly in the scientific literature. Consequently, and as this becomes increasingly easy with electronic health records, we do encourage clinical readers to make such analyses available. However, as with several related diseases (e.g. literature21,24–28), our role as systems biologists is to put together a coherent, systems biology picture, and with all the evidence that we shall review below, it is very clear indeed that RA is driven by a microbial component. To this end, one very major (and in our view clear) differentiator between women and men is the equally more common, anatomy-based prevalence in women over men of urinary tract infection.29–31 This is the first “plank” in Ebringer’s impressive series of arguments (most recently at6,9,32,33) that actually gives a satisfying and coherent account of at least one microbial origin for RA, Proteus spp., that we now review.
Ebringer’s theory (with experiments) that Proteus infection from the urinary tract is a major cause of RA
Ebringer sets his work in a Popperian framework (for reviews of that see literature34–36) but we consider that it is more conveniently set in a systems biology manner as a logical series or chain of intellectually linked events, and this is what we do here. Ebringer (we like especially Ebringer et al.
32
) makes the following 10 claims (the many supporting references are in the paper):
HLA-DR4 lymphocytes injected into a rabbit evoke specific antibodies against Proteus cells (mainly the species mirabilis and vulgaris). Antibodies to Proteus spp. are present in RA patients from 14 different countries. Antibodies to Proteus bacteria in RA patients are disease-specific since no such antibodies are found in other conditions. When RA patients have high titers of antibodies to Proteus such bacteria are found in urinary cultures. Only Proteus bacteria and no other microbes evoke significantly elevated antibodies in RA patients (this is not 100% true, see below). A “shared epitope” EQR(K)RAA shows “molecular mimicry” with the related sequence ESRRAL found in Proteus hemolysis. Proteus urease contains a sequence IRRET which has “molecular mimicry” with the related LRREI found in collagen XI of hyaline cartilage. Sera obtained from RA patients have cytopathic properties against sheep red cells coated with the cross-reacting EQR(K)RAA and LRREI self-antigen peptides. Proteus sequences in hemolysin and urease as well as the self-antigens, HLA-DR1/4 and collagen XI, each contain an arginine doublet, thereby providing a substrate for peptidyl arginine deiminase (PAD) to give rise to citrulline, which is the main antigenic component of CCP, antibodies to which are found in early cases of RA. Antibodies to Proteus come not only from sequences cross reacting to self-antigens but also from non-cross reacting sequences, thereby indicating that active RA patients have been exposed to infection by Proteus.
Some other prokaryotic microorganisms besides Proteus spp. that have been implicated in RA
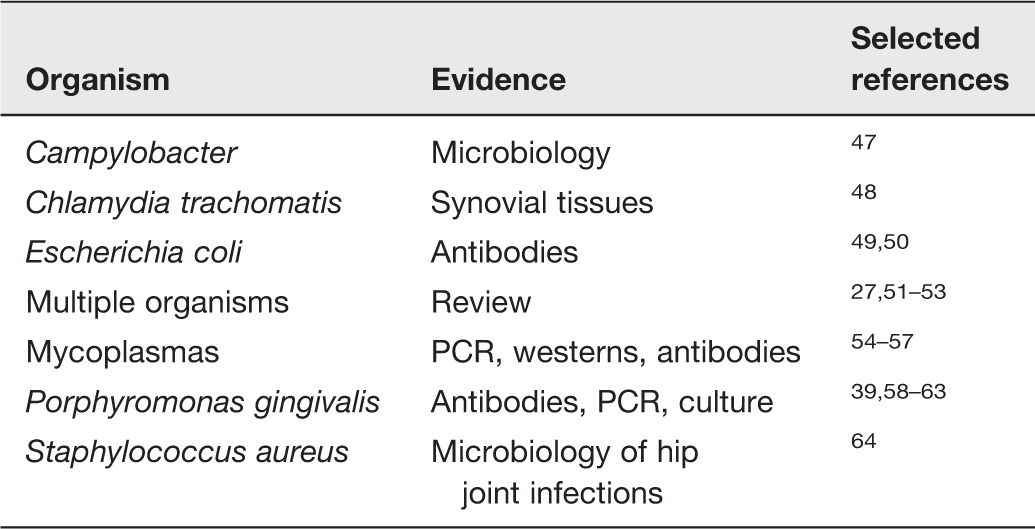
This was a binary (presence/absence) assessment of the microbial contribution, but microbes have many properties beyond presence and absence. In particular, a notable and missing feature of most of these studies (including those of Ebringer) involves (i) the physiological state of the organisms in question, and (ii) what causes them to manifest their activities periodically (for instance as the “flares” characteristic of RA). This we therefore discuss next.
Dormancy, resuscitation, and iron dysregulation
Clinical or infection microbiologists typically recognize bacteria as being in one of two physiological macrostates, either being “alive” (on the basis of their being capable of replication, e.g. to form a colony on a petri dish containing a suitable solid medium), or if not being so capable then being assumed “dead.” However, these are not the only two major physiological states, and indeed they are probably the least common in natural environments! Importantly, the definition of these states is operational: the appearance of a microbe’s physiology also depends on the experiment being used to test it and is not of itself an “innate property” of the organism.65–67 In environmental microbiology, most microbes are non-growing because they lack the nutrients and/or signaling molecules to replicate, but they are not (irreversibly) “dead.” They are best described as “dormant,” and the means by which they are brought back from an apparent state of non-aliveness to one in which they can be cultivated is conventionally known as “resuscitation.” We demonstrated this in a series of papers in laboratory cultures of various actinobacteria (e.g. literature65,68–73), leading to the discovery of an autologous “wake-up” molecule, the “resuscitation promotion factor” or Rpf74–78 that was necessary for resuscitating dormant bacteria in the presence of weak nutrient broth. Note that assays must be done under conditions of dilution to extinction,66,67 to avoid the possibility of simple regrowth of small numbers of cells that were always “alive” and never dormant.
In clinical microbiology, the terms “persister” and “persistent” are commonly used to refer to a phenotypically non-growing (but non-dead) subset of microbes, typically those that have been treated with but tolerant to antibiotics (e.g. literature27,28,79–86). In general terms, these too are operationally dormant as defined above, though their relative physiological states (e.g. as judged by expression profiles) are not really established. We have recently summarized the evidence for a dormant blood microbiome including in red cells21,27,28 (and see also Damgaard et al. 87 ) to complement other literature pertaining to white cells and tissues (e.g. literature88–92). Such dormant cells are, of course, well placed to create inflammation via a continuing shedding of inflammatory agents such as LPS and molecules with antigenic properties as described above. We note that the emergence of infection may also accompany treatment of the disease,93–95 although whether this is due to resuscitation (as we suspect 96 ) or reinfection is not yet uniformly clear.
The question then arises as to what kinds of stimuli trigger the resuscitation. Two are well established. One is the stimulation of Gram-negative bacterial growth by the stress hormone noradrenaline (NA) and other auto-inducers.97–103 One of the roles of NA is to act as a siderophore,104,105 since it is normally available iron that limits microbial replication in vivo,106–111 a phenomenon that adds considerably to the undesirable and purely chemical effects of the second one, which is the presence and availability of unliganded iron that is liable to catalyze the highly damaging Fenton reaction25,112 (see Figure 2 adapted from Kell et al.
27
).
A bacterial system contains distinct subpopulations (1), that we classify as culturable, dormant and non-culturable (2). Specific attention is given to persister cells (3), and the inter-relationship (4) between the subpopulations and phenotypic switching between culturability and dormancy (5). Throughout we follow a systems biology approach to suggest resuscitation due to various triggers like iron and noradrenaline (6). (A color version of this figure is available in the online journal.)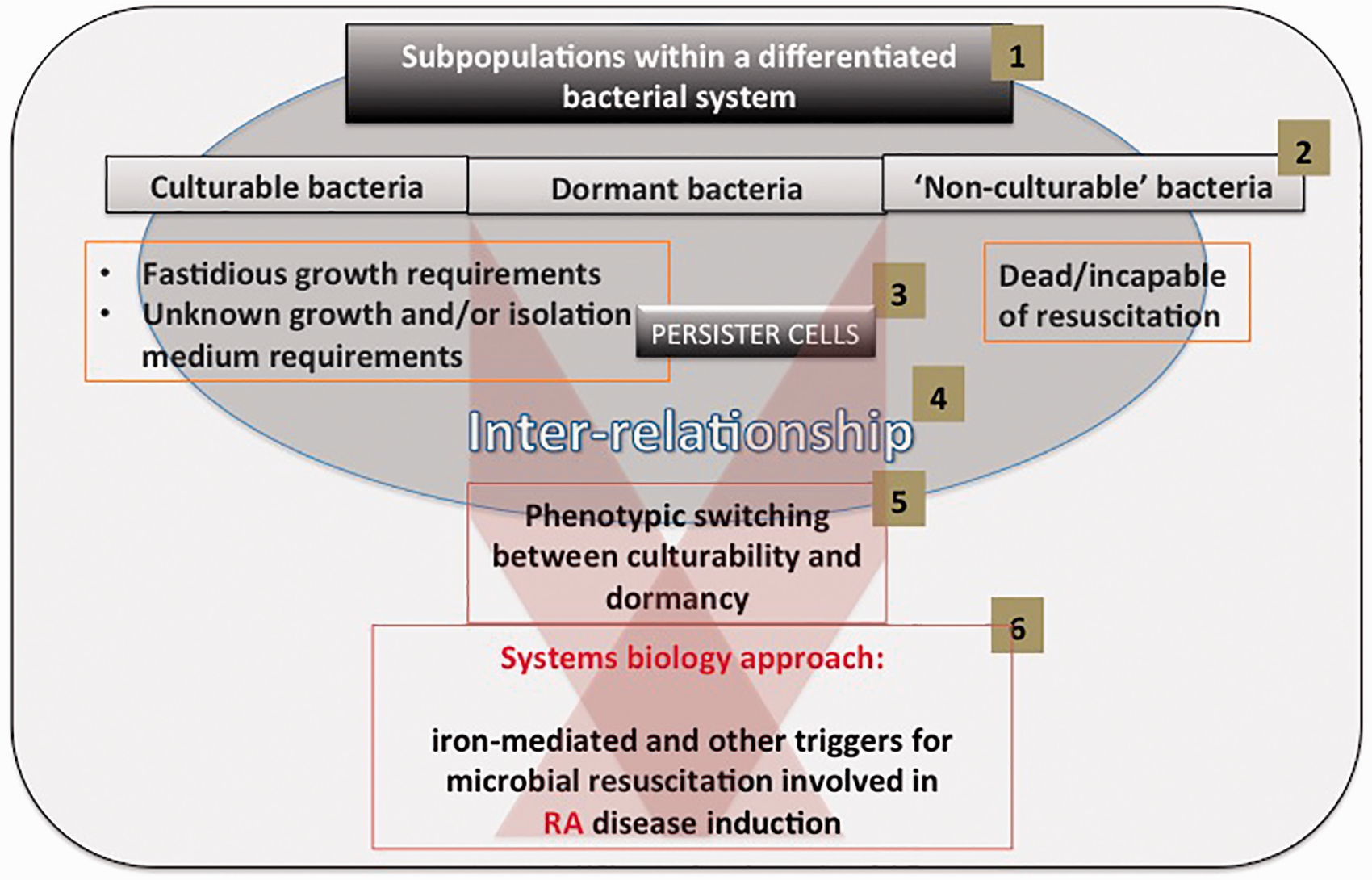
Pathophysiologic markers of inflammation in RA
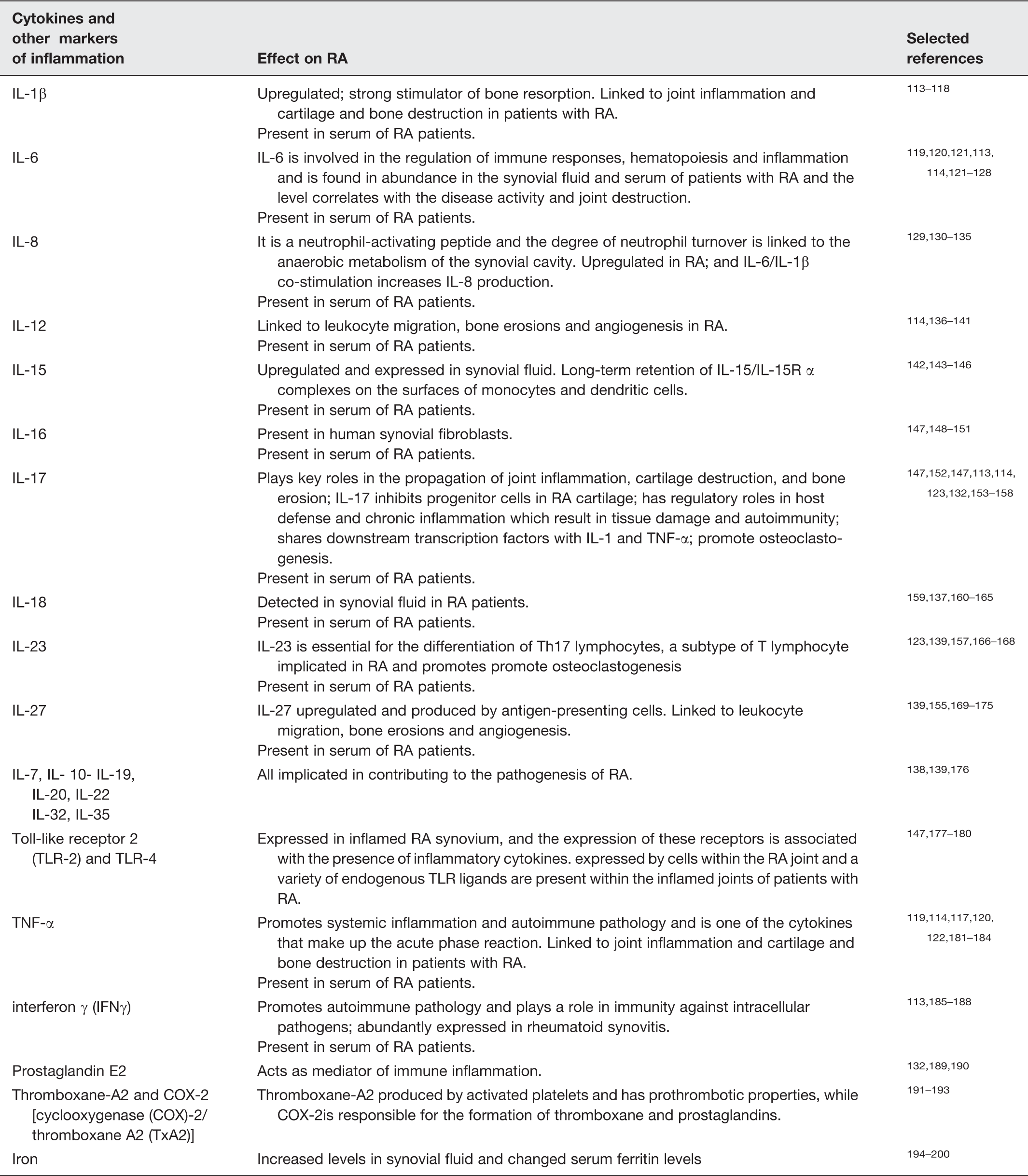
Dysregulated inflammatory markers in Rheumatoid arthritis
Systemic inflammation, cardiovascular disease, and RA
An analysis of the co-existing conditions of rheumatoid arthritis patients patients from a rheumatoid arthritis clinic in South Africa, showing baseline demographics of subjects (n = 38) with Rheumatoid Arthritis
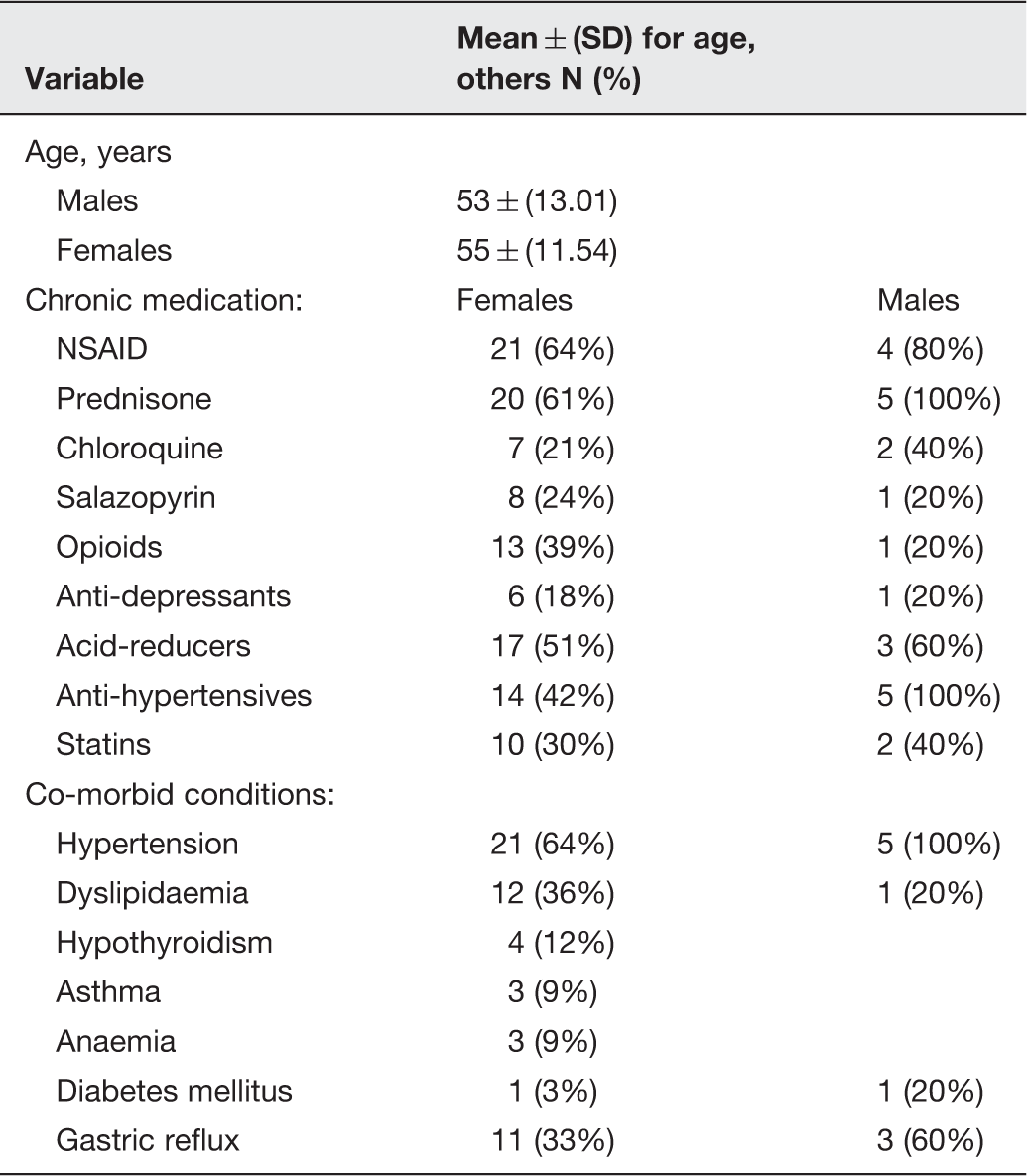
Cardiovascular comorbidities relate more than others to disease activity in RA, and particularly type 2 diabetes and hyperlipidemia were found to be associated with disease activity. 211 Cardiovascular disease, and particularly diabetes, has also been linked to gut dysbioses and bacterial translocation.212–216 Recently, it was also suggested that a co-morbidity index should be used both at baseline, and as a continuous variable in analyses in RA patients, 217 as some co-morbidities are causally associated with RA and many others are related to its treatment. 218
Systemic inflammation is entirely consistent with the plethora of diseases and comorbidities that exhibit iron dysregulation,24,112 raised serum ferritin, 219 hypercoagulation and hypofibrinolysis, 25 and anomalous morphological changes in both erythrocytes and fibrin.220,221 We thus turn to hypercoagulation in RA.
Hypercoagulation, erythrocyte (RBC), and platelet involvement in RA as a result of systemic inflammation
Another hallmark of systemic inflammation (as well as cardiovascular pathology) is clot hypercoagulability, and a hypercoagulable state is also found in RA,222–231 together with a decreased clot lysis ability,25,225,226 possibly due to genuine amyloid formation. 232 Systemic inflammation, oxidative stress, and hypercoagualability all affect erythrocytes (RBCs) and platelets. One of the changes due to the systemic inflammation and oxidative stress noticeable in RBCs and platelets is the production of cell-derived microparticles (MPs). 219
Flow cytometry is the usual method to quantify MP233,234; unfortunately, the small size of these structures and lack of standardization in methodology complicates measurement. 235 Mostly, as MP contain surface and cytoplasmic contents of the parent cells and bear phosphatidylserine, antibodies to specific cell surface markers and annexin V can be used for identification235,236 or for tissue factor-dependent FXa generation assays. 237 Their sizes can vary but are of the order of 50 to 800 nm 238 and are therefore easily detected on and around their mother-cell by scanning electron microscopy. 209
In RA, circulating MPs exposing complement components or activator molecules are elevated, 239 and their levels are correlated with disease activity. 240 These MPs are also of great important in cardiovascular diseases, and this may be one reason for the enhanced cardiovascular morbidity and mortality seen in RA. 240 Furthermore, MPs may contribute to the local hypercoagulation and fibrin deposition in inflamed joints of patients with RA. 241
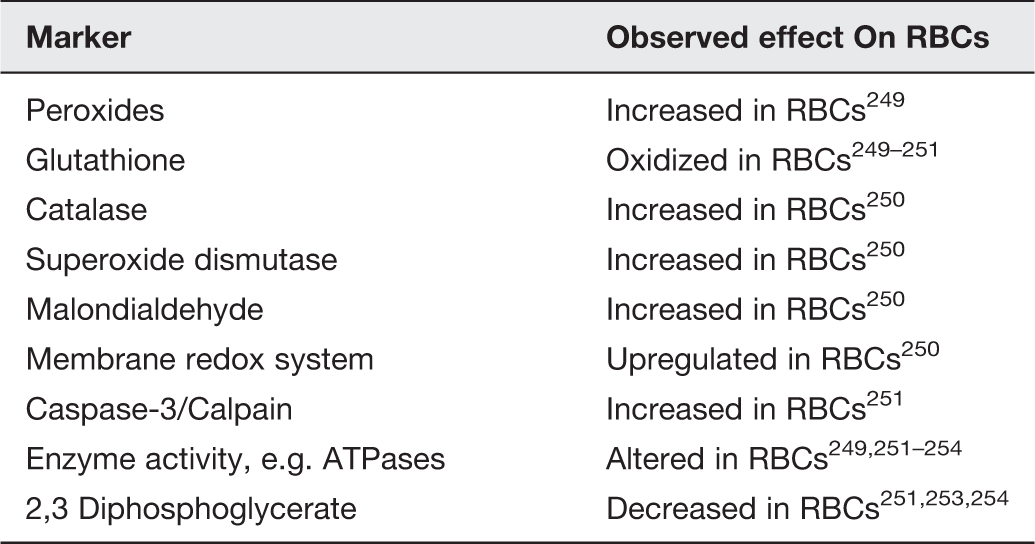
Markers taken as indicators of oxidative stress in RBCs
Pathologic changes in coagulation result directly in abnormal fibrin fiber formation during clotting, and MPs associated with platelets, as well as membrane changes of RBCs, can be visualized by scanning electron microscopy in blood smears from RA individuals. See Figure 3(a) for an example of healthy fibrin fiber formation versus pathological fibrin fiber formation in RA (Figure 3(b)), and a healthy platelet showing a clear cell body and pseudopodia formation with a smooth membrane (Figure 4(a)) versus platelets from RA where activation, spreading, and MP formation are visible (Figure 4(b) and (c)). Red arrows in Figure 4(c) possibly indicate round ultramicrobacteria, which differ from the much more irregularly shaped MPs.
Fibrin fiber formation in the presence of thrombin (a) healthy fibrin and (b) rheumatoid arthritis fibrin with matted fibrin. Scale: 1 µm (a) A healthy platelet with prominent cell body and pseudopodia formation and smooth membrane; (b) two spreaded and activated platelets with microparticle formation (irregularly shaped structures closely associated with membranes (white arrows)) in rheumatoid arthritis; red arrows showing much rounder structures – possibly ultrabacteria; (c) a higher magnification of an RA platelet with microparticles budding off spreaded platelet. The scale bars are (a): 200 nm; (b): 1 µm and (c): 200 nm. (A color version of this figure is available in the online journal.)
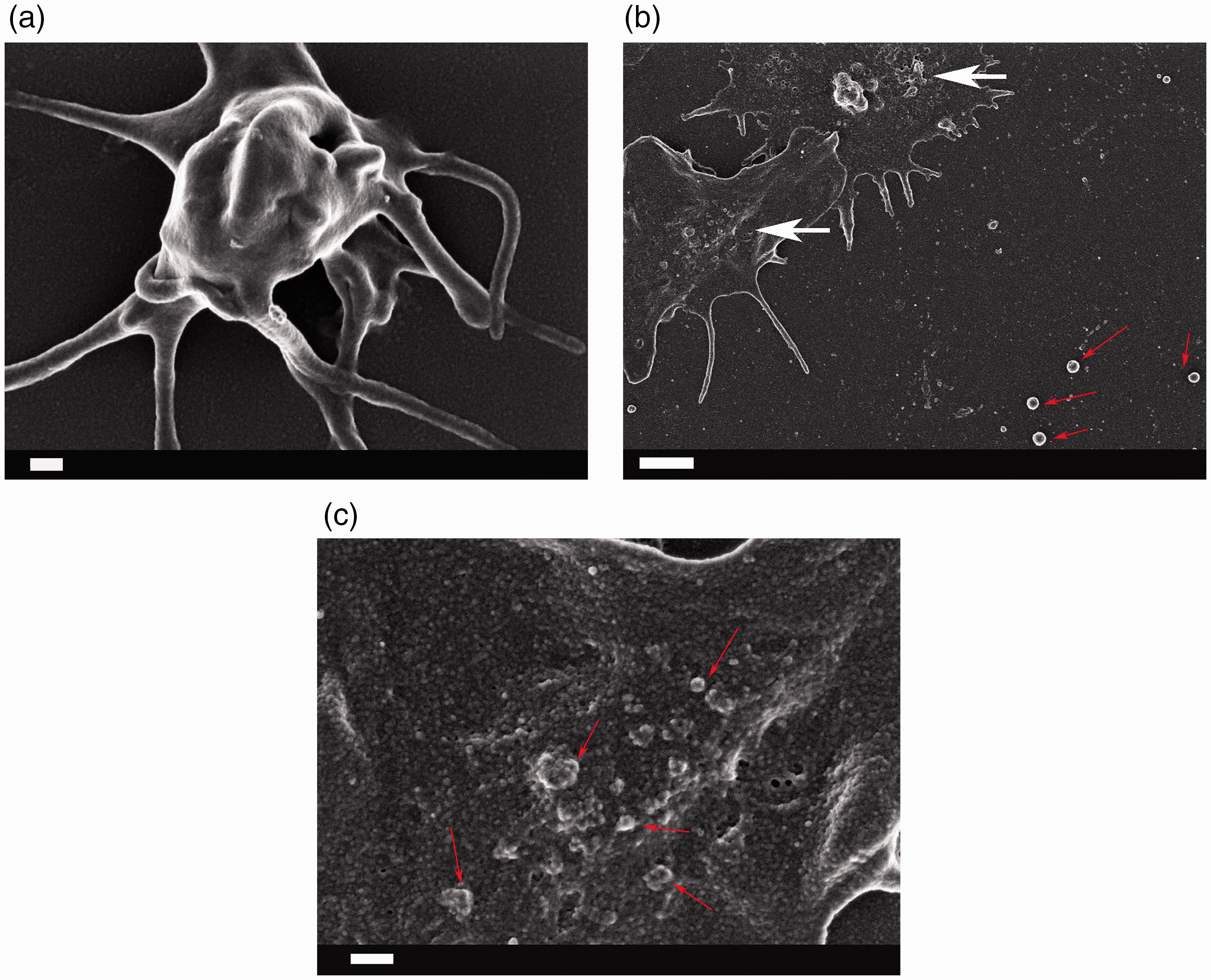
Figure 5(a) and (b) shows a typical healthy RBC with no membrane changes, compared to normal RBCs, of a typical RA individual (Figure 5(c) and (d)), where RBC folding due to loss of structural integrity is seen in the presence of fibrin fiber formation (when thrombin is added (Figure 5(c))) and membrane changes are observed with MP formation (Figure 5(d)). The changed membrane surface is also confirmed via roughness analysis of RBC membranes using atomic force microscopy (AFM), where a significantly increased roughness was noted in RA RBCs compared to the case of healthy RBCs (Figure 6).
(a) and (b) A representative healthy RBC (b is higher magnification showing the membrane); (c and d) A representative rheumatoid arthritis RBC with folding (c) and visible membrane microparticle formation. Scale bars for (a) and (c): 1 µm; scale bars for (b) and (d): 100 nm RBC membrane roughness analysis as seen with atomic force microscopy (AFM) of a representative micrograph from a healthy RBC (a) and a rheumatoid arthritis RBC (b). (A color version of this figure is available in the online journal.)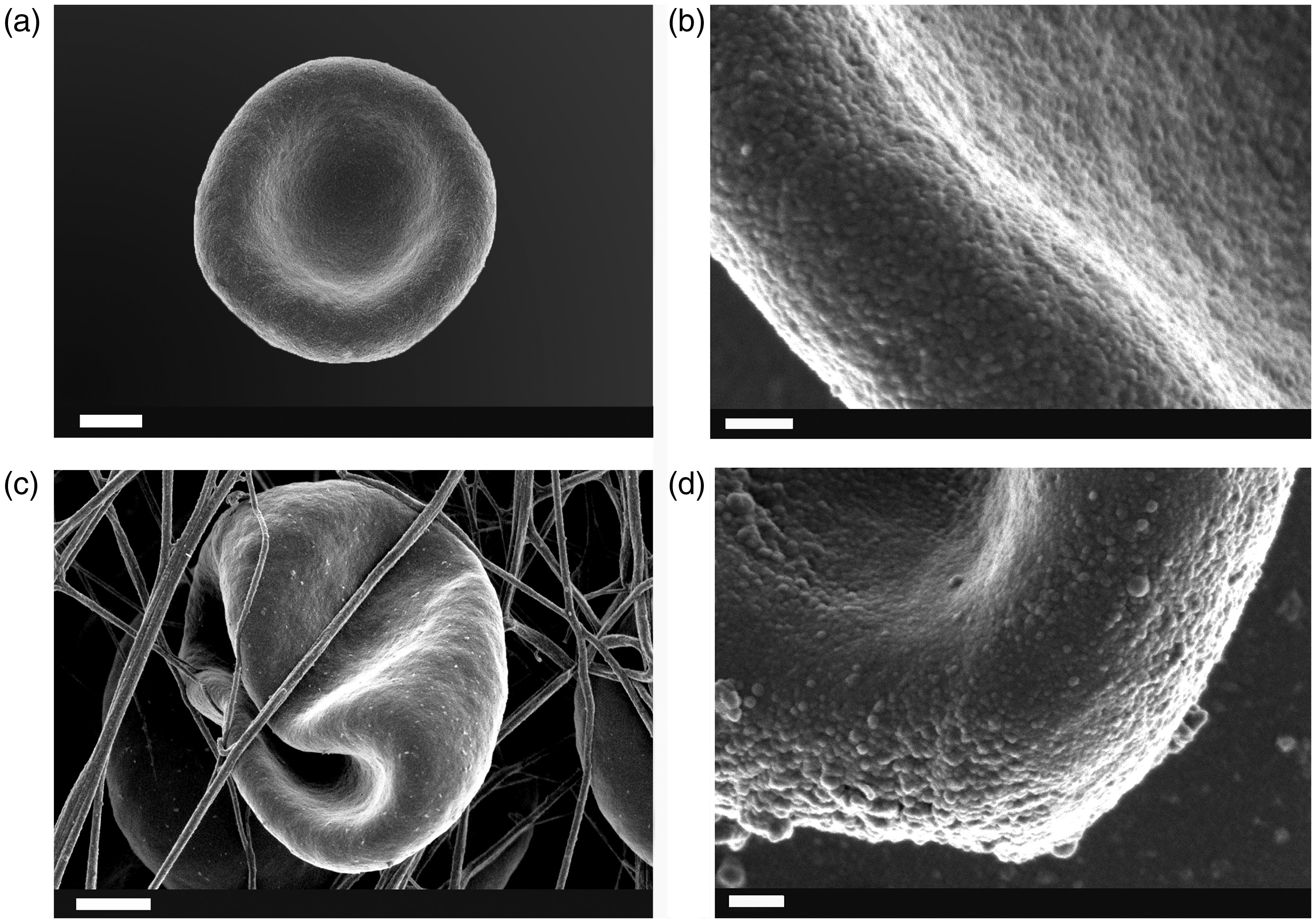

A note on amyloidogenesis and amyloidoses in RA
In a recent development,232,255,256 we have shown that the fibrin dense-matted deposits of the type shown in Figure 4(b) can be generated by tiny amounts of LPS, and that they are actually β-amyloid in nature. This has significant implication more generally, 257 including diseases such as sepsis 258 and pre-eclampsia. 259 Amyloid fibrils of various kinds are widely recognized as inflammatory and cytotoxic (e.g. literature260–263). We do not yet know the origins of the amyloid seen in RA, but it has long been known that amyloidoses are an important accompaniment (and potentially exacerbator) of RA (e.g. literature264–268).
Present treatments treat the symptoms
There are two main pharmacological treatments for RA, viz. small molecule disease-modifying anti-rheumatic drugs (so-called sDMARDs) and biologicals (bDMARDs). We shall discuss these in general terms, especially for the former from the perspective of potential antibacterial effects. However, we need to start with present metrics for the severity of disease.
Metrics of disease severity
Given that there seems little general recognition of the real causes of RA, and that the symptoms including pain can be very severe, it is understandable, as this overall section implies, that many treatments are aimed at alleviating the symptoms. We also recognize that at least some of those that “work,” may do so, if inadvertently, by treating the causes as well. But to begin with, we need to consider the metrics of disease severity in common use.
A great many approaches to assessing disease severity exist, and the tendency to prescribe more drugs to those with more severe disease is necessarily 269 a confounding factor. 270 Databases for medical claims around disease severity have value, 270 but they tend to lack information on important clinical variables, such as the number of tender and swollen joints, which would traditionally be used to assess disease severity in RA. 271 Where comparisons exist, the information regarding, e.g. the presence of swollen joints and disease severity, are not well correlated. Objective measures of variables such as inflammatory cytokines are attractive, but for the patient, the severity, especially of pain, is subjective, and patient-assessed severity indices are consequently common. This said, the patient global assessment (PGA) 272 is noteworthy 273 as while using seven objective criteria based on stiffness and swelling, pain (being presumably too subjective) is not among them. Other studies did find some correlations between swelling, stiffness, and pain of joints.274,275
The European League Against Rheumatism (EULAR) has a number of useful recommendations, including treatment/management, 276 efficacy,277,278 and joint imaging. 279 For our present purposes, these will suffice entirely.
Small molecules (sDMARDs) and their role(s) as antibacterials
The chief recommendations 276 are to start early and monitor frequently. These are seen as the strategy of first resort, with methrotrexate, sulfasalazine, and leflunomide being seen as front line drugs (possibly along with low-dose glucocorticoids). It is highly noteworthy that sulfasalazine is in fact an antibiotic (one of the first), as it is split in the intestine into aspirin and the antibiotic sulphapyridine,280,281 while methotrexate shows antibiotic activity against organisms as diverse as S. aureus 282 and Plasmodium vivax. 283 As mentioned above, “first treatments” are the least likely to be confounded by bias occasioned by the fact that later treatments will be favored by patients with more severe or refractory disease, a phenomenon that probably confounded a study of minocycline and doxycycline. 284 However, and while – like other drugs285,286 – they probably have multiple effects, a good many studies indicate the utility of the antibiotics minocycline and doxycycline in treating RA (e.g. literature287–294). After sDMARDs have been tried, the recommendation 276 is to move to a biological.
Biologicals (bDMARDs)
The chief biologicals276,277,295–298 are inhibitors of TNF-α, including monoclonals, and inhibitors of the IL-6 receptor; they all decrease inflammatory symptoms, and it is unclear whether they might have any direct or indirect antibacterial effects. Their chief issue is that, as proteins, they can themselves cause (auto) antigens to be raised,299–301 while, as mentioned above, any dampening of immune system response may increase the likelihood of novel or emergent infection.
Iron chelation as a therapeutic?
If the “dormant microbial” hypothesis is correct, it is to be predicted that nutritional and/or pharmacological iron chelators would be of clinical benefit. Although we are not aware of any clinical trials to this end, a number of papers indicate the therapeutic benefits of chelators in RA (e.g. literature302–305 Iron chelation may also have a useful mechanistic role in iron redistribution.306–308 Thus, as with Alzheimer’s disease, where the established benefits 309 of iron chelation languished for decades,310,311 it would seem that iron withholding (e.g. literature312,313) might be an important therapeutic strategy in RA that is well worth exploring.
In a similar vein, we would predict or anticipate a great preponderance of RA in those with iron overload diseases, as indeed seems to be the case.314,315
A role for lipopolysacharides (LPS) in RA
Recently, we summarized the evidence for a significant involvement of lipopolysaccharide shed by dormant and resuscitating bacteria as underpinning the chronic inflammation characteristic of a variety of diseases, and suggested that LPS may play a role in the pathogenesis of RA. 21 The presence and role of LPS may be supported by a recent review that provided evidence for the involvement of a microbiome in inflammatory arthritis and rheumatic diseases. 316 Recently, Scher et al. 317 also found strong correlates between the presence of Prevotella copri and new-onset untreated RA patients.
Certainly, LPS is also known to upregulate all of the cytokines upregulated in RA and mentioned in Table 1. In our recent review, 21 we also focused on the fact that antibodies could be generated to LPS that – like the anti-Proteus antibodies mentioned in detail above – might also serve as autoantibodies of significance in RA and in particular during the flares (that may be ascribed to periods of particular resuscitation activity).
The generalized LPS also exerts its effects via activation of cytokines such as IL-6, and TNF-α in response to LPS,
115
IL-8,
318
IL-12,
319
IL-15,
320
thereby exciting the innate immune response. The scheme is typically as follows (extensively discussed in Kell and Pretorius
21
):
Production of a variety of pro-inflammatory cytokines,326–328 where NF-κB plays a prominent role329,330 via a set of canonical pathways illustrated in Figure 7. NF-κB translocates to the nucleus to turn on a great many genes in a frequency-dependent fashion, including in particular TNF-α and IL-6.331–333 At high concentrations of LPS,334,335 it also activates a “non-canonical” inflammasome pathway, which is independent of TLR4336,337 (see Figure 8).
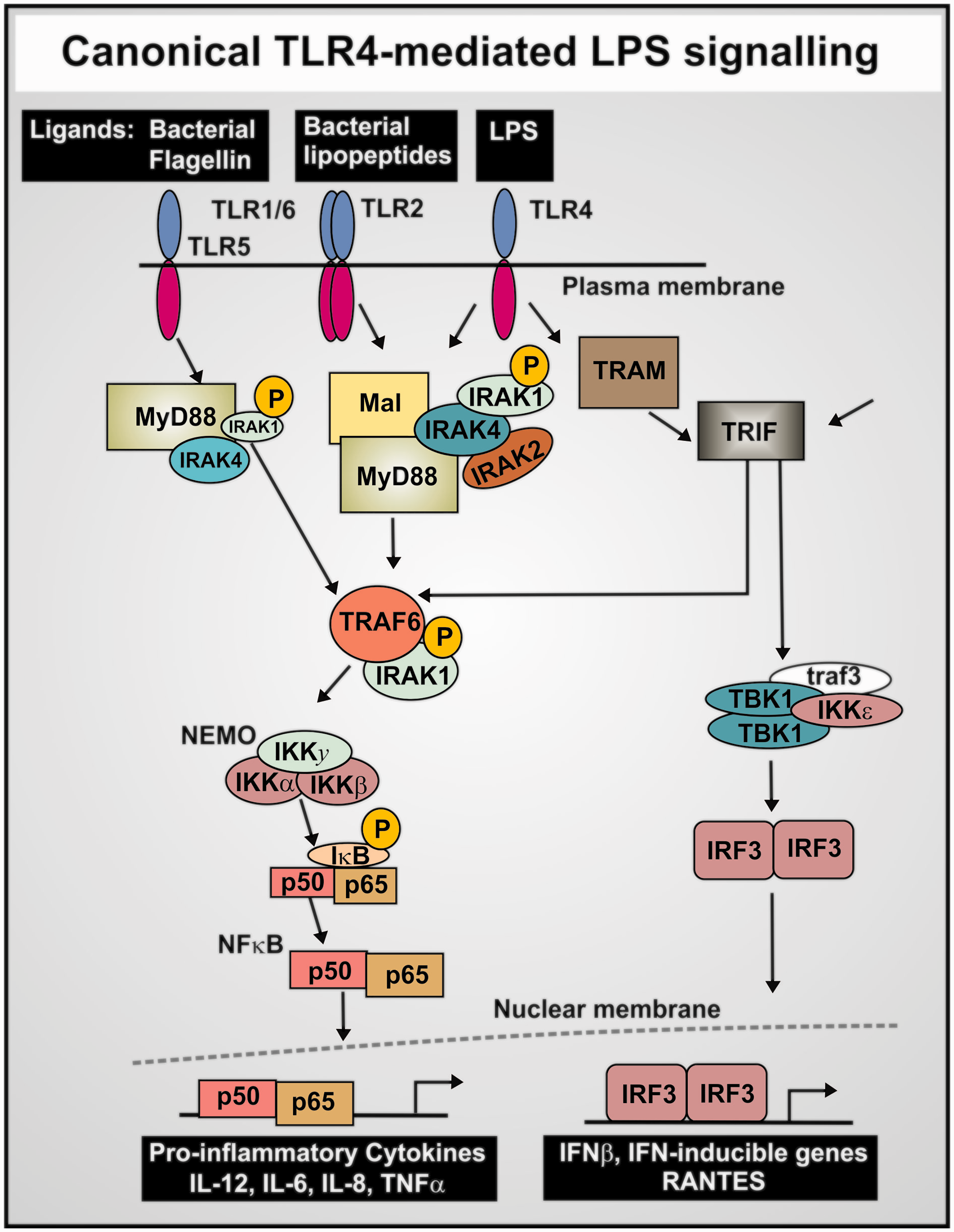
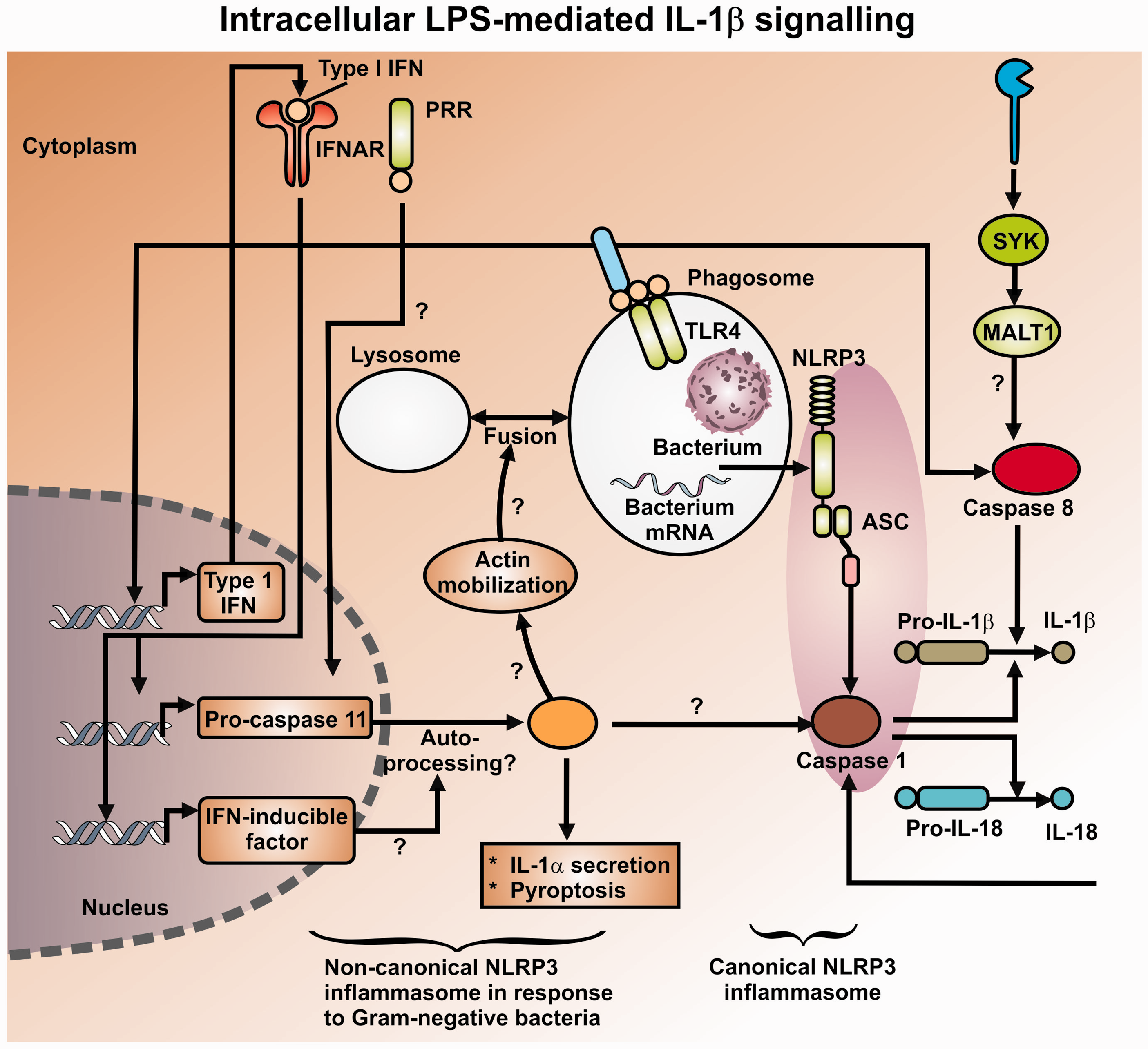
Finally (see above), LPS may catalyze the formation of inflammatory and cytotoxic β-amyloids. Consequently, we might again suggest that appropriate antibacterials and iron chelators (that can hitchhike on the necessary transporters282,339–343) would serve to lower this stimulus, and in contrast to the biologics actually strike at the root causes of the disease.
Further tests of our hypothesis
While we have adduced much evidence in favor of the view that recurring infection by (resuscitating dormant) bacteria is in fact a, if not the, major and ultimate cause of RA, albeit there is not a unitary “cause,” our views do come with multiple predictions that may be tested (of course some have been already, see above, in many cases extensively).
Bacteria should be detectable in relevant tissues of RA patients, whether by culture or by molecular methods (e.g. macromolecular sequencing or antibodies). Relevant products such as LPS and other antigens should be detectable in patients vs. controls. Their numbers (bacteria and/or inflammatory products) should increase with disease severity and during “flares.” Their numbers and activity (hence disease prevalence/severity) should correlate with free iron levels. Treatments that lower the activity of bacteria and/or their products should be of significant therapeutic benefit. These may include iron withholding, antibacterial, anti-LPS, and anti-amyloid treatments.
Although probably not yet seen as mainstream, a number of therapeutic strategies based on these and other ideas (including the roles of vitamin D metabolites, that for reasons of space we do not discuss here) do indeed seem to have enjoyed success (e.g. literature344–348).
Concluding remarks
As mentioned previously, 27 while it can be difficult (but cf 349 ) to ascribe causality in complex biochemical networks, an accepted strategy within the philosophy of science, that rather accurately describes the systems biology approach, is to the effect that if a series of ostensibly unrelated findings are brought together into a self-consistent narrative, that narrative is thereby strengthened. This is known as “coherence.”350–353 We have sought, we believe successfully, to bring together the evidence for a coherent narrative that links infection, microbial dormancy, iron dysregulation, and inflammation as part of the etiology of RA. Importantly, the proposals can easily be tested further, both diagnostically and therapeutically.
Footnotes
Authors’ contributions
All authors participated in the design, interpretation of the studies, and analysis of the data and review of the manuscript. EP wrote the paper and prepared images; DBK wrote and edited the paper; OA prepared samples: PS identified patients, collected samples and verified the clinical relevance.
Acknowledgments
We thank the National Research Foundation (NRF) and Medical Research Council (MRC) of South Africa for supporting this collaboration. This article is paper 10 in the series “The dormant blood microbiome in chronic, inflammatory disease.”
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Ethical approval was granted at the University of Pretoria (Human Ethics Committee: Faculty of Health Sciences): E Pretorius.
Funding
This work was also a contribution from the Manchester Centre for Synthetic Biology of Fine and Speciality Chemicals (SYNBIOCHEM) (BBSRC grant BB/M017702/1).