
Abstract
We describe a clinical, imaging and biomarker phenotype associated with an amyloid precursor gene (APP) E665D variant in a 45-year-old man with progressive cognitive and behavioral dysfunction. Brain MRI showed bilateral, confluent T2 hyperintensities predominantly in the anterior white matter. Amyloid imaging and CSF testing were consistent with amyloid deposition. 7 Tesla MRI revealed cerebral microhemorrhages suggestive of cerebral amyloid angiopathy (CAA). Contrary to previous reports, this case raises the possibility that the APP E665D genetic change may be pathogenic, particularly given the abnormal Alzheimer’s disease biomarkers observed in the cerebrospinal fluid, positive amyloid imaging and imaging evidence for CAA in a relatively young patient with progressive cognitive decline.
Keywords
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia. It is a leading cause of disability in the elderly and as life expectancy continues to rise so will the incidence and prevalence of this disease. Early onset AD (EOAD), defined as disease onset before the age of 65 years, has been a focus of research to help elucidate the pathophysiology of this complex condition. Mutations in three genes including amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) genes account for approximately 11% of EOAD. 1 These genetic cases produce a wide variety of atypical clinical phenotypes including spastic paraparesis, myoclonus, seizures and cerebral amyloid angiopathy (CAA) with intracerebral hemorrhage. 2
The APP gene is located on the long arm of chromosome 21. The APP gene appears to play an important role in synaptic activity and neuronal plasticity, although its functions are not well understood. 3 The APP gene was first suspected to be biologically linked to AD given the near universal presence of AD pathology in older individuals with Down’s syndrome. 4 Pathological APP mutations cause AD or CAA, while several missense variants have been described as non-pathogenic or are of unclear pathogenicity. For example, the APP E665D missense variant located on exon 16, has been described previously as non-pathogenic. 5 Over thirty APP mutations have been identified within exons 16 or 17. 6,7 These mutations are associated with dementia and AD pathology including amyloid deposition, neurofibrillary tangle formation and CAA.
EOAD has also been reported in several different families with a variety of APP mutations including the Flemish Ala692Gly variant, 8 Arctic Glu693Gly mutation 9 and the Iowa Asp694Asn mutation. 10 The common clinical characteristics associated with these APP mutations include progressive cognitive decline, CAA, myoclonus, and seizures. 11,12 This case report describes a clinical phenotype associated with an APP genetic variant previously thought to be non-pathogenic. 5
Case Report
A 45-year-old Caucasian male with a history of hypertension, gout and alcohol abuse presented to our clinic with cognitive dysfunction. His symptoms started in his mid-thirties. He owned and operated a small business and noted difficulty with short term memory, multi-tasking, along with visuospatial deficits. He was forced to keep a detailed calendar with his daily activities though co-workers still noted a decline in his overall work performance. He also endorsed impaired word retrieval and phonemic paraphasias. He was admitted to our in-patient epilepsy unit after several episodes of confusion and disorientation for continuous EEG monitoring at the age of 42. Testing revealed intermittent, rhythmic, generalized slowing on the EEG tracing without epileptic activity. During that admission, his metabolic panel was unremarkable outside of mildly elevated aspartate transaminase 65 U/L (reference range 7-40 U/L) and alanine transaminase 61 (reference range 5-50 U/L) thought to be related to his alcohol use. HIV, tuberculosis, hepatitis and syphilis testing were negative and his vitamin B1, B6, B12 and thyroid stimulating hormone were within normal limits.
The patient had two MRIs at 1.5 Tesla, spaced nearly three years apart (at the age of 42 and 45, respectively). Both scans showed bilateral, confluent T2 white matter hyperintensities, predominantly in the anterior frontal and temporal white matter, which were essentially stable over the three-year span (Figure 1). There was no evidence for gradient echo sequence (GRE) abnormalities on the 1.5 Tesla MRIs to suggest evidence for cerebral microhemorrhages or intracranial hemorrhage. The more sensitive susceptibility imaging on the 7 Tesla MRI revealed multiple, small foci of microhemorrhages on susceptibility weighted imaging (SWI), almost all located within the cortical ribbon, suggestive of CAA (Figure 2). There were no SWI changes within the basal ganglia, thalamus, or pons to suggest they were hypertensive in etiology. There was no evidence for an intralesional central vein on the 7 Tesla MRI that is frequently detected in multiple sclerosis. 13 There was no abnormal enhancement in the brain. MRI of the cervical, thoracic and lumbar spine were unremarkable without abnormal signal or enhancement. Initial CSF testing during his index epilepsy admission was normal without evidence for pleocytosis or intrathecal antibody production.

1.5 Tesla MRI FLAIR images illustrating signal abnormality predominantly in the anterior white matter which extends into the anterior temporal lobes. There is sparing of the corpus callosum, brainstem and cerebellum. Overall, his MRI shows relative stability over the three-year follow-up period. FLAIR; Fluid Attenuation Inversion Recovery.

Amyloid PET ([18F]florbetapir) imaging demonstrates amyloid deposition with corresponding, co-registered MRI and CT imaging. From left to right: amyloid PET, 1.5 Tesla FLAIR; CT; and amyloid PET imaging. The orange arrows indicate areas positive for abnormal cortical amyloid primarily within the occipital lobes. The yellow arrows show regions of reduced amyloid radiotracer uptake which correspond to the white matter disease seen on the MRI/CT scans. The bottom row (box B) are 7 Tesla MRI susceptibility weighted images illustrating scattered cortical lesions (red arrows) consistent with cerebral amyloid angiopathy. PET, positron emission tomography; FLAIR; Fluid Attenuation Inversion Recovery; CT, computerized tomography.
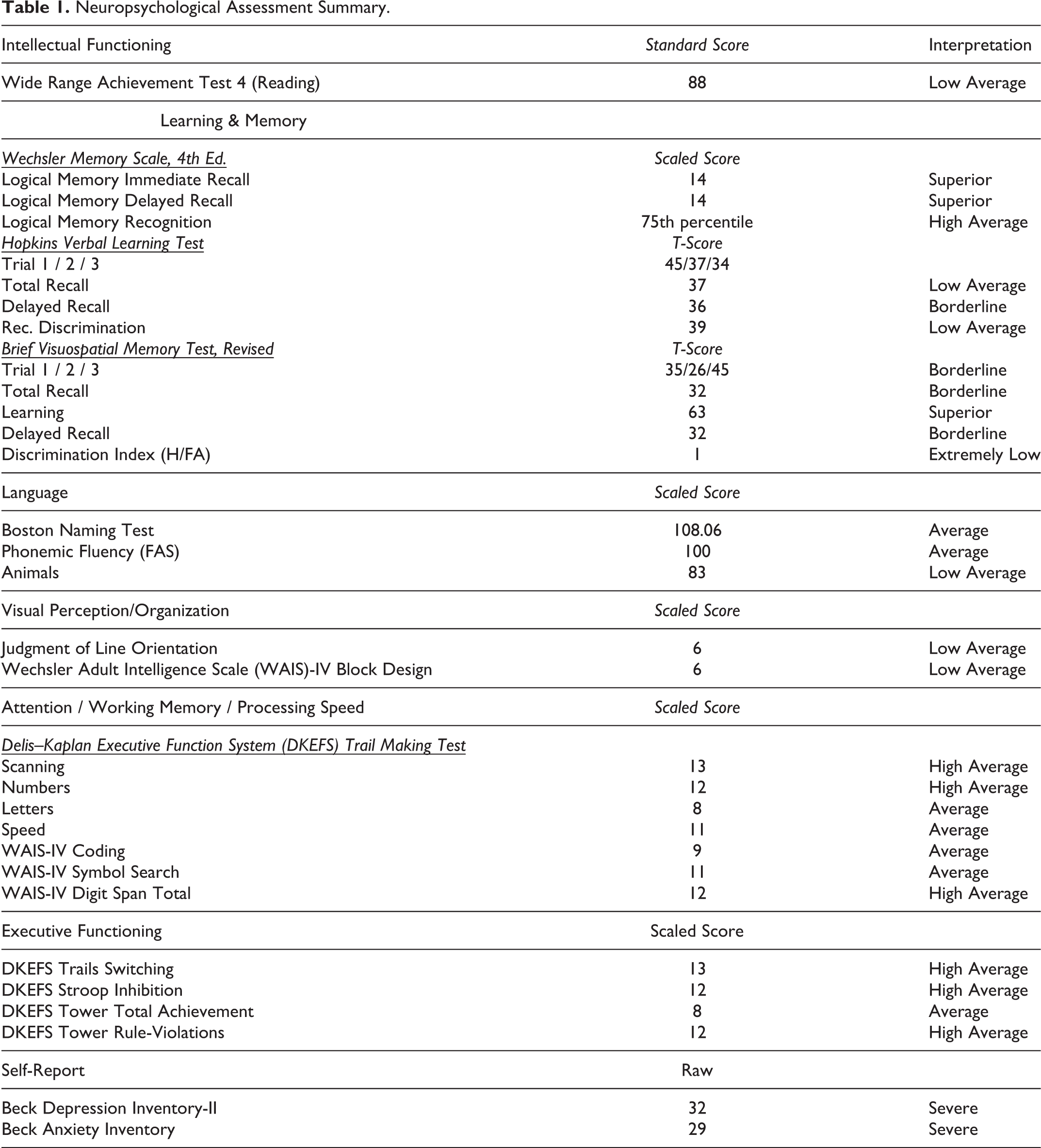
During his initial evaluation in our clinic, we performed a detailed neurological examination along with neuropsychological testing. The patient graduated from the 10th grade but required speech therapy in kindergarten and additional math tutoring in fourth grade. His baseline level of cognitive function was estimated to be low-average to average. Formal neuropsychological testing revealed significant variability in memory performance along with mild inefficiencies in aspects of executive functioning (inhibition), semantic fluency, and visuospatial function (Table 1). Overall, his testing suggested cognitive deficits localized to the subcortical regions and was consistent with the MRI findings. His remaining neurologic examination was significant for mildly dysmorphic facial features including hypertelorism and low set ears, mild incoordination, symmetric hyporeflexia and reduced vibration sense in the lower extremities. His EMG was normal and did not reveal evidence of a generalized sensorimotor polyneuropathy.
Neuropsychological Assessment Summary.
A genetics consultation was placed given the concern for hereditary leukodystrophy. A 3-generation pedigree was performed (Figure 3). The patient had four full siblings. His two brothers had undifferentiated, medically intractable epilepsy complicated by global developmental delay. The patient also had two sisters without any pertinent medical history. The patient had three daughters. The eldest was in her early twenties with a history of attention-deficit/hyperactivity disorder (ADHD) but there was no history of cognitive impairment. His remaining daughters were diagnosed with autism and ADHD. Extensive efforts to retrieve the other family members’ medical records, or neuroimaging results were unsuccessful.

Family pedigree with the proband identified by the arrow. SIDS, sudden infant death syndrome; d, died.
Further serum evaluation including lactate, pyruvate, very long chain fatty acids, arylsulfatase A, plasma amino acids, carnitine, urine oligosaccharides were normal. His lysosomal 1,4-alpha-glucan-branching enzyme activity was also normal. A leukodystrophy gene panel did not show any significant findings (Medical Neurogenetics, LLC, Atlanta GA). Whole exome sequencing was performed and illustrated two variants of unknown clinical significance without evidence for a Notch3 gene mutation (GeneDx, Gaithersburg, MD). 14 The two variants were the APP p.E665D (c.1995G>C) and myosin heavy chain gene (MYH)-6 p.G572 R (c.1714G>A). The Bekris laboratory confirmed the presence of the APP mutation using targeted sequencing in exon 16 (E665D; rs63750363) by directly sequencing PCR genomic DNA products utilizing methodology previously described. 15,16 Of note, the MYH variant has been associated with hypertrophic as well as dilated cardiomyopathies, although his extensive cardiac evaluation did not reveal any abnormalities. Human apolipoprotein E (APOE) status, determined by allelic discrimination analysis of whole blood genomic DNA, 17 revealed APOE ε2/ε4 alleles.
In order to assess for potential AD biomarkers within the CSF, two independent laboratory evaluations were completed. The first was through Athena Diagnostics (Marlborough, MA) using ELISA kits (Innotest β-amyloid [1-42], Innotest hTAU-Ag, Innotest Phospho-Tau[181P], Innogenetics, Ghent, Belgium). 18 This testing showed low amyloid-ß42 (Aß42) 335.6 pg/mL (normal reference range > 500 pg/mL) and elevated total-tau 395.9 pg/mL (normal reference range < 350 pg/mL) and phosphorylated-tau 62.6 pg/mL (normal reference range < 54 pg/mL). 19,20 The Aβ42/t-tau index (ATI, calculated as Aβ42/(240 + 1.18 t-tau) was 0.47 which was “consistent with AD” based on Athena Diagnostics reference range (AD reference range ATI < 0.8).
A separate analysis by the Bekris laboratory was performed using the Luminex 200 3.1 xPONENT System and MILLIPLEX® MAP Human Amyloid Beta and Tau Panel (EMD Millipore, Chicago, IL, USA). Using this method, CSF amyloid-ß40 (Aß40) was 2041 pg/mL and Aß42 183.69 pg/mL with an Aß42/40 ratio of 0.09 which was consistent with AD. 21,22
Amyloid positron emission tomography (PET; [18F]Florbetapir (Amyvid, Eli Lilly, USA) imaging confirmed amyloid deposition (Figure 2), with the greatest deposition in the occipital lobes. The pattern of amyloid tracer accumulation was unusual compared with typical cases of late onset AD due to the reduced radiotracer uptake within the subcortical white matter lesions compared to the adjacent cortex (Figure 2; yellow arrows). This pattern of reduced radiotracer binding has been previously reported in demyelinating lesions 23 and non-specific white matter hyperintensities 24 though the exact mechanism for this failure has not been fully elucidated.
Discussion
This case describes a potentially new clinical phenotype associated with an APP variant previously thought to be non-pathogenic. 5 Our patient reported progressive cognitive decline with objective deficits in multiple cognitive domains. His MRI scans showed symmetric, predominantly anterior white matter signal abnormality, which was initially concerning for a leukodystrophy. However, a comprehensive leukodystrophy evaluation including a gene panel was unrevealing. In addition, his 7 Tesla MRI showed evidence for CAA. His work-up eventually illustrated an APP E665D variant, along with APOE ε2/ε4 alleles, which prompted an evaluation for AD. Amyloid deposition was confirmed with CSF biomarkers and amyloid PET imaging. His family history suggested a potentially hereditable, neuropsychiatric condition. Unfortunately, those conditions were not well classified and it is difficult to draw any specific conclusions from his pedigree.
It is difficult to make a formal diagnosis of CAA in this case. The widely accepted Boston diagnostic criteria for CAA is applied in patients over the age of 55 with 1.5 Tesla MRI. 25,26 Our lower strength 1.5 Tesla GRE scans did not identify any abnormalities consistent with CAA. In addition, application of the Boston criteria in a population of patients with the hereditary APP Dutch type mutation 27 found a diagnostic sensitivity of 48 percent with a particular poor performance within asymptomatic patients. 28 His APOE genotype is another confounding factor. His APOE ε2/ε4 status is relatively uncommon in the general population and is not well studied in patients younger than 40 years old making it difficult to comment on its role in this specific case. 29 A single APOE ε2 allele is considered to be a protective against AD. 30 That being said, it is unclear whether the protective effects of APOE ε2 outweigh the APOE ε4 increased susceptibility to AD. 30 In one study, older patients with APOE ε2/ε4 alleles had an odds ratio of developing AD of 2.6 (95% CI 1.6-4.0) 31 which has been confirmed in a more recent study. 32 APOE status may have less impact in EOAD including those with Down’s syndrome. 29,33 Overall, it is possible that the presence of ε4 carrier status contributed to the pathogenicity of the APP genetic change.
A previous APP variant at 665 codon which resulted in the same substitution of aspartate for glutamate (Glu665ASP: E665D) has been reported in the literature. 5 The proband was an 86-year-old female who developed late-onset dementia along with intermittent episodes of confusion. At autopsy, she fulfilled the neuropathological criteria of AD. Her son also developed early onset dementia, but had multiple vascular risk factors and did not have the APP E665D mutation. It was thought his decline was more likely related to his multiple ischemic strokes as he was diagnosed with vascular dementia. 5 The variant was not found in 40 control subjects or 127 dementia patients. Given the conflicting results, the authors were unable to conclude that this variant was pathogenic 5 and to our knowledge, there have been no further reports describing the pathogenicity of this mutation. This study was published in 1994 and APOE genotype and markers for amyloid deposition were either not available or performed. Thus, it is not possible to comment on whether there was asymptomatic deposition of β-amyloid. In addition, the report did not mention MRI findings to determine if there was wide spread white matter changes similar to our case.
The majority of reported APP mutations involve the β- and γ-secretase cleavage sites which are utilized in the normal metabolism of the APP transmembrane protein. One of the first reported mutations in the APP gene was the “London” APP V717I mutation. This clinical phenotype featured prominent cognitive decline, dyscalculia, and prominent myoclonus and seizures. 12 Other mutations in this domain can have early, predominate cerebrovascular complication. This can be complicated by severe CAA leading to recurrent cerebral hemorrhage. 34 -36 The fact that our patient had evidence for CAA on his 7 T MRI supports the notion that the E665D APP variant is pathogenic. 37
The APP E665D variant is very conservative in terms of amino acid sequence and possibly structure, resulting in one less methylene group in the amino acid side chain, which otherwise still bears the carboxylic acid at its terminus. While this conservative mutation might have little or no effect on the protein structure or function, the possibility of a significant biological effect cannot be excluded. Overall, we do have strong evidence for amyloid deposition in our patient. This includes CSF testing utilizing two methods, positive amyloid PET imaging and 7 Tesla MRI findings suggesting CAA. Whether these changes account for his clinical presentation is less clear. His vascular risk factors and continued alcohol abuse obviously confound his clinical course. Overall, it is impossible to determine what role those factors played in his cognitive dysfunction but neither of those factors accounted for the amyloid deposition or MRI findings. Genetic information on other family members was not available which also limits our interpretation.
Although this case does not conclusively identify this APP E665D variant as pathogenic for causing cognitive dysfunction, we can more conclusively state that it likely contributed to his amyloid deposition given the previously described APP mutations in AD pathology. Overall, it is important to continue to explore the wide spectrum of clinical symptoms associated with APP abnormalities.
Footnotes
Abbreviations
Alzheimer s disease (AD); early onset AD (EOAD); amyloid precursor protein (APP); presenilin 1 (PSEN1); presenilin 2 (PSEN2); cerebral amyloid angiopathy (CAA); gradient echo sequences (GRE); susceptibility weighted image (SWI); attention-deficit/hyperactivity disorder (ADHD); amyloid beta (β); myosin heavy chain gene (MYH); human apolipoprotein E (APOE); attention-deficit/hyperactivity disorder (ADHD); positron emission tomography (PET).
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article. JRA: nothing to disclose. SEJ: Siemens and Monteris (speakers fee and travel). Biogen MS-PATHS research support. Advisory board for Monteris and Eisai. ML: Research support from NIH. LMB: nothing to disclose. MK: nothing to disclose. KK: nothing to disclose.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: JBL: Received research funding from the Alzheimer’s Drug Discovery Foundation, Douglas Herthel DVM Memorial Research Fund, Eisai, GE Healthcare, Jane and Lee Seidman Fund, Lewy Body Dementia Association, Michael J Fox Foundation, National Institute of Health (P30 AG062428, UO1 NS100610; R13 NS111954), and Sanofi. He has received consulting fees from Acadia, Biogen, Eisai, and Takeda.
Significance Statement
– Amyloid precursor protein mutations are commonly associated with progressive cognitive decline and cerebral amyloid angiopathy.
– This case outlines a clinical phenotype associated with an amyloid precursor protein variant (APP E665D) that includes progressive cognitive decline, extensive white matter changes and findings suggestive of cerebral amyloid angiopathy. Contrary to prior reports, this case suggests this APP variant may be pathogenic due to the age of symptom onset, confirmed amyloid deposition and family history.
Statement of Informed Consent
Written informed consent for patient information and images to be published was provided by the patient.