
Abstract
Background:
Subcortical vascular dementia relates to small-vessel disease and hypoperfusion, resulting in focal and diffuse ischemic white matter lesions. The main target of the disease are the frontal subcortical neural networks. There is no clinical standard definition of the pathology, on the contrary, everyday clinical practice suggests dominant behavioral alterations and dysexecutive syndrome.
Methods:
The aim of this study was to investigate gait disorders, behavioral alteration, and drug intake of a subcortical population with dementia (n = 1155). A complete neuropsychological examination was conducted at baseline and every 6 months, and the results were compared.
Results:
Our data suggest that there is a significant increment in apathy levels and a dramatic decrease in gait and equilibrium control in the patients examined during follow-up.
Conclusion:
Subcortical vascular dementia may be associated with gait and balance alteration and apathy per se; we suggest to implement clinical data with these major aspects.
Introduction
Besides strategic infarct dementia and intracranial hematomas, dementia caused by cerebrovascular disease (commonly referred to as “vascular dementia” [VaD]) is often associated with subcortical ischemic vascular lesions caused by small-vessel disease. Subcortical vascular dementia (sVaD) now incorporates the old entities “lacunar state” and “Binswanger’s disease” and relates to small-vessel disease and hypoperfusion, resulting in focal and diffuse ischemic white matter lesions and incomplete ischemic injury.
Patients with sVaD are believed to represent a prevalent and homogeneous group. Characteristic features of the condition include a subcortical syndrome, comprising progressive cognitive impairment with frontal features and dysexecutive syndrome, and mood and personality changes such as depression and emotional lability. According to National Institute of Neurological Disorders and Stroke and Association Internationale pour la Recherché et l’Enseignement en Neurosciences (NINDS-AIREN), sVaD is mainly due to lacunar infarct occurring in distribution of small arterioles, usually in the white matter, basal ganglia (BG), thalamus and pons, or to microinfarct not seen on macroscopic examination, small area of cystic, or noncystic necrosis surrounded by astrocytes. 1 Incomplete infarct may also be present, due to a selective loss of neurons, myelin, and oligodendrocytes, without cystic necrosis, occurring in the periphery of major artery distribution infarcts (eg, penumbra) or in deep white matter. Incomplete white matter infarcts are associated with myelin pallor, astrocytosis, and a variable degree of axonal loss. 2
Although gait and equilibrium are fundamental patterns of everyday life and functional independence measures, these 2 important symptoms have not been widely considered. Well-accepted studies suggest and confirm that leukoaraiosis is associated with gait impairment, worse equilibrium scores, and falls. 3 -8 Some authors argue these gait disorders may exist in mild subclinical forms associated with mild periventricular changes. A severe gait disorder clearly occurs in Binswanger’s disease in association with a severe leukoaraiosis. 9
It has been recently stated that gait disorders are common in the elderly patients and are related to loss of functional independence and death. Cerebral small-vessel disease is related to gait disturbances: in particular, both white matter lesions and lacunar infarcts were independently associated with most gait parameters, especially with stride length. These authors concluded that white matter lesions in the BG, internal capsule, and limbic areas and lacunar infarcts in the frontal lobe and thalamus were related to a lower velocity. 10 Recently, a direct relationship between subcortical vascular lesions and falls has been established, even if works on the matter are scarce. 11 -13
On the other hand, the clinical significance of negative symptoms such as apathy is increasingly recognized in neurological and psychiatric disorders, particularly those associated with frontal-subcortical dysfunction.
14,15
Apathy is defined as lack of motivation, manifested by diminished goal-directed behavior, reduced goal-directed cognition, decreased emotional engagement, a reduced interest and participation in normal purposeful behavior, problems in beginning or sustaining an activity, lack of concern or indifference, and a flattening of affect. The prevalence of apathy in neurodegenerative disorders, such as Parkinson’s disease (PD), vary between 16.5% and 51%, depending on the instrument for assessment and on the samples examined. Apathy is quite common also in sVAD; different studies try to define its role in Alzheimer’s disease (AD), but even the most recent and well-conducted ones did not distinguish between early and advanced stages of AD or even between AD and AD with parkinsonism.
14,16-17
It has been hypothesized that dysfunction of the nigrostriatal pathway may play an important role in the pathophysiology of apathy in neurodegenerative disorders. In fact, apathy seems to be independent of disease duration, disability and severity of parkinsonism, and
We tried to define gait, balance and equilibrium alterations, and a behavioral complex symptom such as apathy in a well-defined population, having sVaD (standing from a neurological, clinical, and radiological criteria), considering even precipitant factors, such as concomitant pathologies and consequent therapies. We now present an extension of the work, with a speculation on what we observed in a 5-year follow-up.
Methods
Study patients were men and women aged 68 to 94 years, entering in Cognitive Disorder Unit Evaluation of the University of Trieste, with Mini-Mental State Examination scores 24 of at least 14 and satisfying the fourth edition of the Diagnostic and Statistical Manual of Mental Disorders for dementia, 25 recruited from June 1, 2007, to June 1, 2013. We have examined 1648 patients with VaD, in particular 1243 patients with sVaD. Among them, 57 died during follow-up and 31 did not have a caregiver who could guarantee adequate compliance and have been excluded from the study. The remaining 1155 patients (566 men and 589 women) satisfied the criteria for probable VaD in accordance with the NINDS-AIREN criteria. 1 A patient was diagnosed as having sVaD when the computed tomography (CT)/magnetic resonance imaging (MRI) scan showed moderate to severe ischemic white matter changes 26 and at least 1 lacunar infarct. Brain CT scans or MRI images were randomized and assessed independently, after the radiologist’s opinion, by a neurologist (RM).
Patients were not included in the study if they showed signs of normal pressure hydrocephalus, previous brain tumors, previous diagnosis of major stroke, or brain hemorrhage. We did not include patients with white matter lesions caused by specific etiologies, such as multiple sclerosis, brain irradiation, collagen vascular disease, and genetic forms of VaD, such as cerebral autosomal dominant arteriopathy or cerebral autosomal recessive arteriopathy. Patients with previous major psychiatric illness (ie, schizophrenia, bipolar disorders, psychosis, compulsive–obsessive disorders, etc) or central nervous system disorders and alcoholism were excluded too. We did not consider depression among exclusion criteria, according to different studies 27 and the potential correlation with VaD predisposing factor.
This was a prospective cohort study, designed to investigate gait (balance and equilibrium) disorders, behavioral alterations, and drug intake of a subcortical dementia population. Study patients were 1155 men and women, aged 68 to 94 years, diagnosed with sVaD, not bedridden, and outpatients who underwent a standardized baseline assessment that included a detailed history, a physical examination, laboratory tests, and psychiatric evaluations. The physical examination included evaluations of pulse rate and rhythm, blood pressure, heart size and sounds, peripheral pulses, retinal vessel and carotid artery evaluation as well as blood pressure measurement in dorsal decubitus and in the orthostatic position, electrocardiographic evaluation, and chest x-ray. The physical examination was repeated at every visit; electrocardiographic evaluation and laboratory tests were repeated every 12 months. Patients were allowed to continue any previous therapy (eg, cholinesterase inhibitors, antihypertensive, antidyslipidemic, and antidiabetic drugs). All patients were followed with periodical neurological and neuropsychological examinations. Visits were scheduled to take place every 4 months.
A complete neuropsychological examination was conducted at baseline and every 6 months; the results at baseline, at 12, at 24, and at 60 monthswere compared. The trial was conducted in accordance with the Declaration of Helsinki and with the Ethics Guidelines of the Institute. Global performance was assessed using the clinical dementia rating (CDR) 28 at every visit. Behavioral symptoms were assessed using the neuropsychiatric inventory (NPI) 29 at every visit. In order to evaluate the apathy as an independent scale, as it is tested along with many other variables in NPI, we employed the Clinical/researcher Rated version of the Apathy Evaluation Scale (AES-C). 30 Functional activities were assessed by the Barthel index (BI), 31 and complex activities of daily living were evaluated by the instrumental activity of daily living. 32 Mobility problems were assessed by the Tinetti scale for equilibrium/balance and gait 33 at every visit. Semiquantitative assessment consisted of the modified Tinetti test with 17 items: 9 for body balance (score 0-16) and 8 for gait (score 0-12) with a maximum score of 28. Hachinski ischemic score was done at every visit. 34 Patients were registered for their medical intake.
Statistical analyses were performed using the SPSS (version 16.0). Within-group changes from baseline to 12, 24, and 60 months were tested using the Wilcoxon signed rank test. This was done for the overall scores for each efficacy variable.
Spearman’s rank correlation analyses were performed between behavioral outcome measures, apathy, Tinetti scale, global, balance, and equilibrium, and BI. Results are presented as mean changes from baseline with standard deviations (SDs), and P values are presented where appropriate.
Results
A total of 1648 patients (789 men and 859 women) were diagnosed as having VaD. The diagnosis was based on historical information and neuropsychological assessment and supported by findings on structural (CT or magnetic resonance) imaging. Subsequent follow-up of patients has reinforced the clinical diagnoses in all cases.
Brain CT scans or MRI images were available for all the 1648 selected patients; 494 patients did MRI studies and 1278 did CT scans. Obviously, the sensitivity for observing subcortical lesions is greater with MRI compared to CT scans. For 1288 patients, CT scans, history, neurological, and neuropsychological tests were sensitive and sufficient to make the diagnosis. In all, 170 patients (not adequate imaging acquisition or not convincing data) completed their diagnostic route with MRI sequences. In all, 190 patients came to our unit with MRI images. Therefore, the patients who did CT/MRI were homogeneously recruited and no demographical, social, cultural, and clinical differences was observed.
A neurologist (RM) revised all the imaging, using the Blennow et al’s 35 scale for CT scans and the Scheltens et al’s 36 scale for MRI imaging. There was 95.8% interrater agreement for the independent assessment of the scans (κ = .8). We excluded from the study the patients diagnosed as having multi-infarct dementia (374) and patients with brain stem infarction (31); finally 1243 patients had sVaD.
Among the 1243 patients with sVAD, 57 died during follow-up and 31 patients did not have a caregiver who could guarantee adequate compliance, and therefore have been excluded after the first year from the study. All the other 1155 patients (566 men and 589 women) completed the full 60-month study. Their mean age was 74.2 ± 5.1 years, and they had a mean education level of 9.7 ± 3.1 years (Table 1).
Baseline Characteristics of Patients.
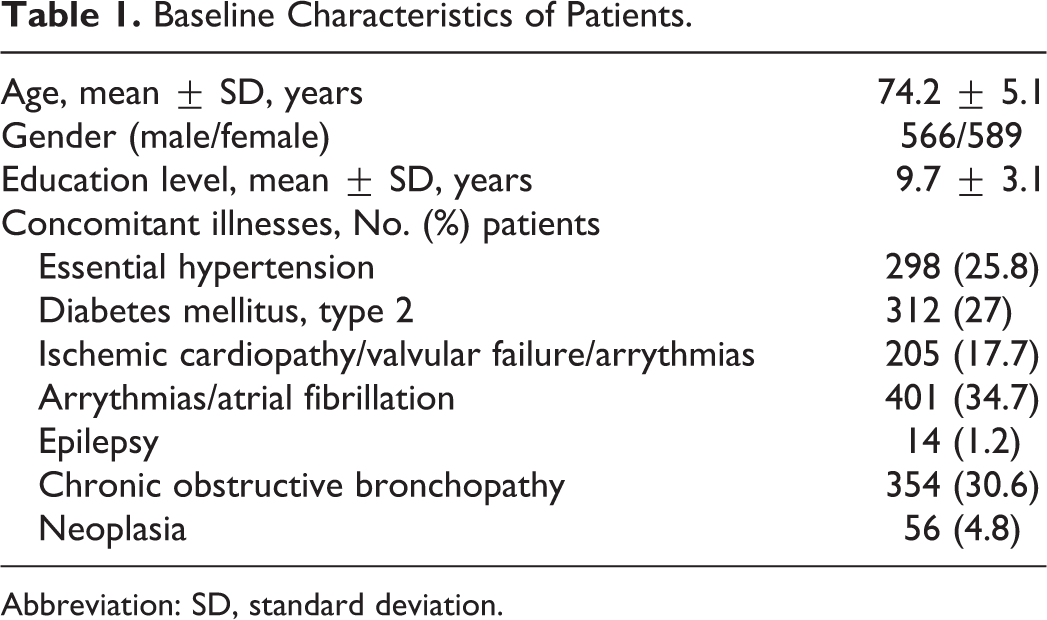
Abbreviation: SD, standard deviation.
Patients were allowed to continue any previous therapy (eg, antihypertensive, antidyslipidemic, and antidiabetic drugs; Table 2). During the follow-up, the patients were prescribed neuroleptics and/or benzodiazepines. Considering the global health conditions, there was an obvious deterioration in the patients as evidenced by the increase in the daily drug uptake (Table 3).
A Synopsis of the Drugs used by the 2 Groups of Patients at Baseline.
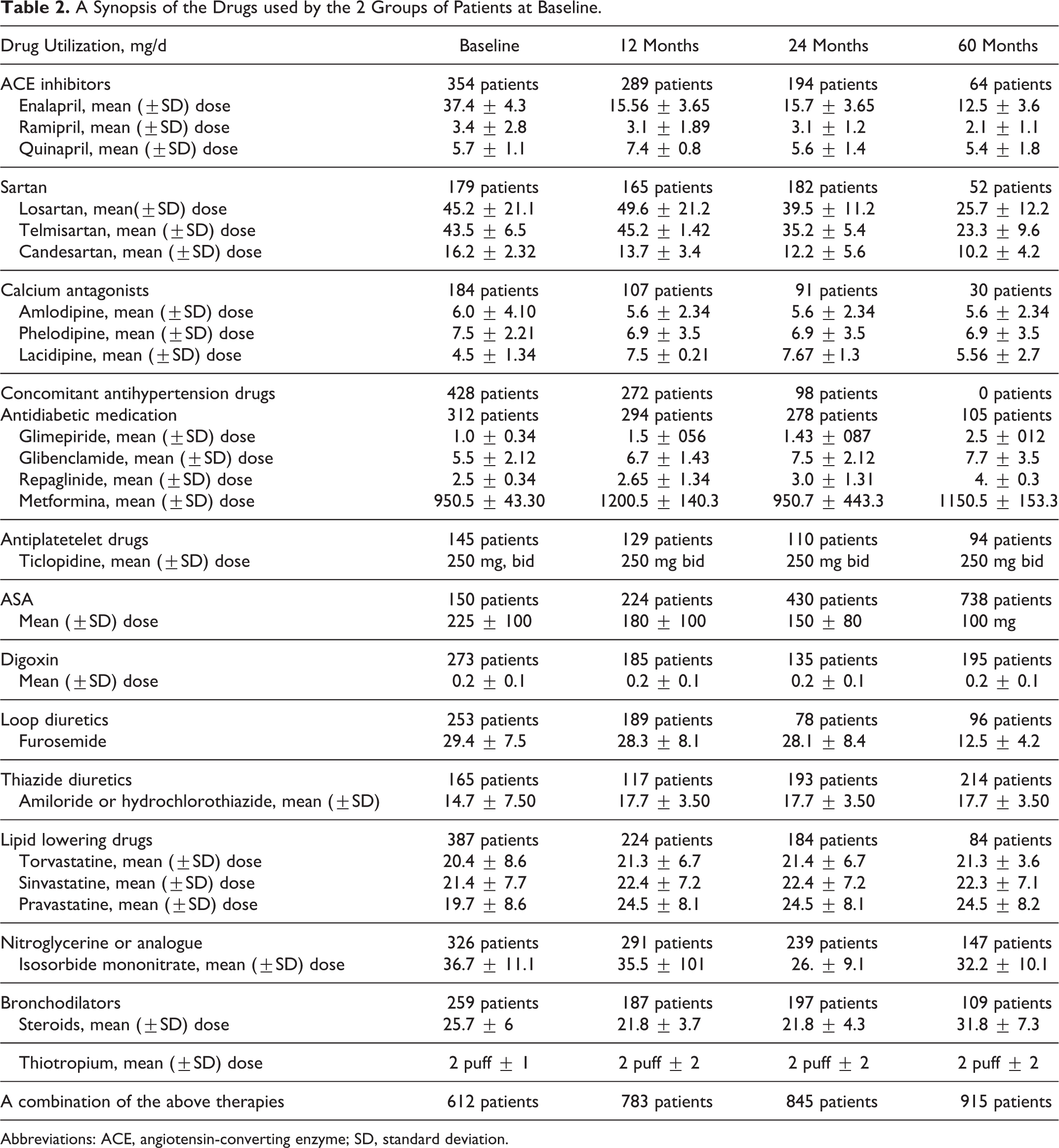
Abbreviations: ACE, angiotensin-converting enzyme; SD, standard deviation.
A Synopsis of the SNC Drugs used by the Patients.
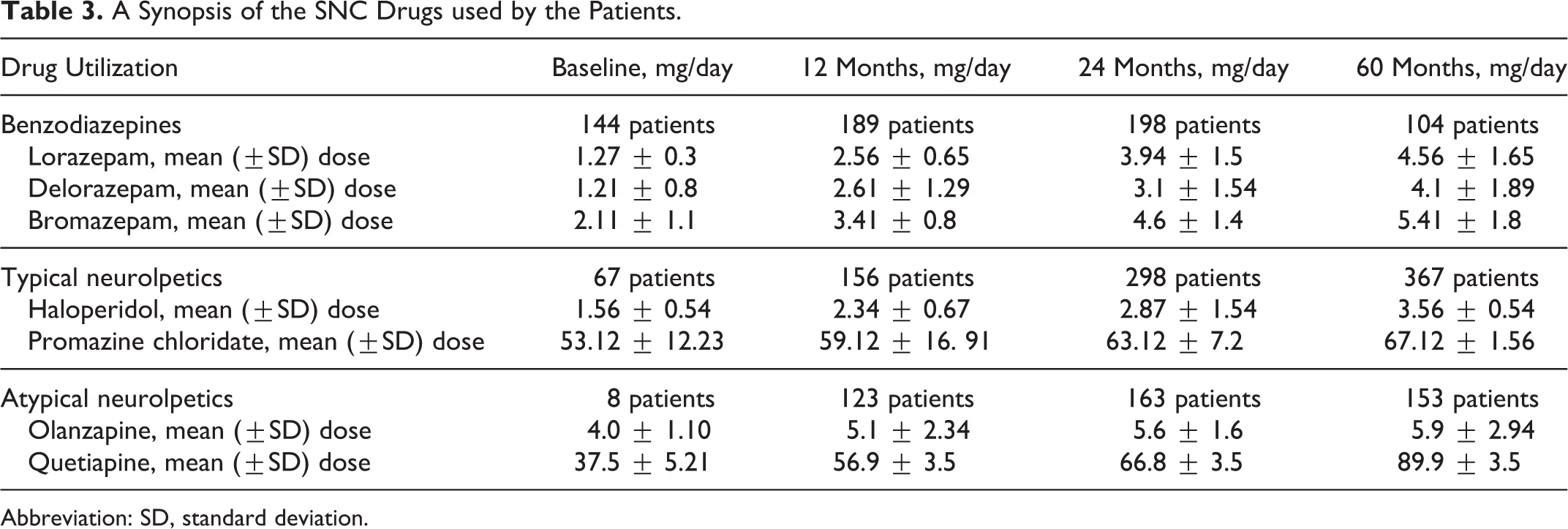
Abbreviation: SD, standard deviation.
Main scores obtained by the patients during the follow-up have been reported in Table 4 as mean and SD. According to a Wilcoxon signed rank test, there was a general significant worsening of the clinical, cognitive, behavioral, and instrumental capacities (a part from the Hachinski scores) of the patients observed in the 60-month follow-up (Table 5). Apathy scores, reported as rough measures, and their evolution during 60-month follow-up have been summarized in Tables 6 and 7. There is a significant increment of apathy levels in the patients examined during follow-up.
Tests Results in the Patients Observed During Follow-Up.
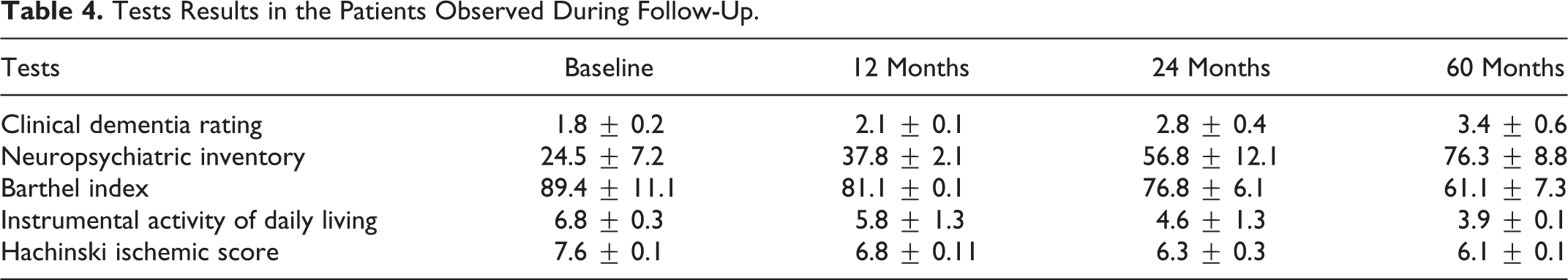
Results at 60 Months: A Comparison Over Baseline, 12, and 24 Months.

Abbreviation: ns, not significant.
Apathy Scale Measured by AES-C Observed During Follow-Up.
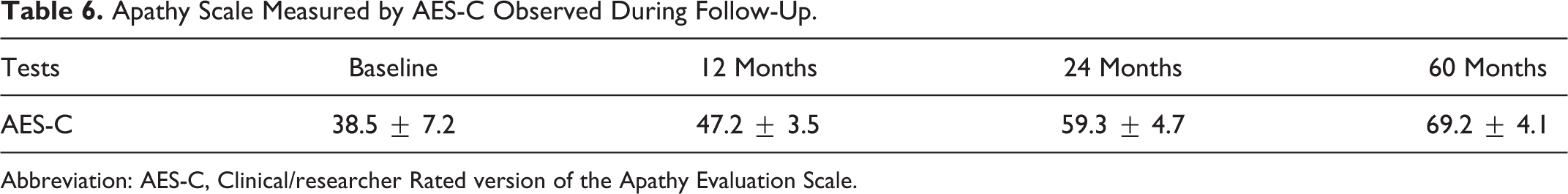
Abbreviation: AES-C, Clinical/researcher Rated version of the Apathy Evaluation Scale.
Apathy Scale Measured by AES-C Results at 60 Months: A Comparison Over Baseline, 12, and 24 Months.

Abbreviation: AES-C, Clinical/researcher Rated version of the Apathy Evaluation Scale.
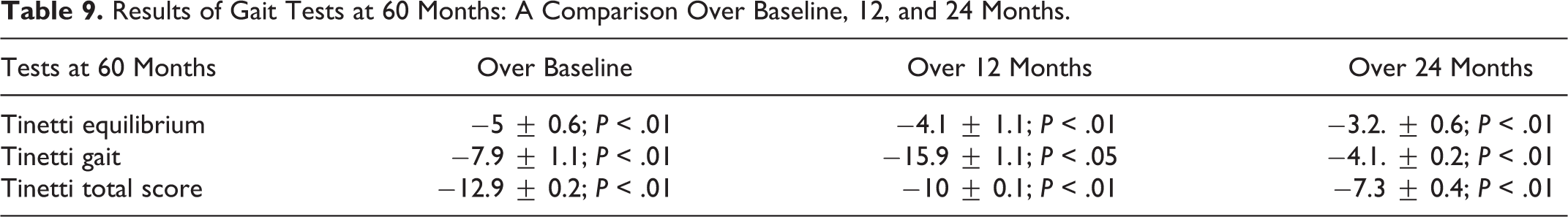
The results obtained in the Tinetti scale evaluation, reported as subscores of gait and equilibrium as said before and the total score, have been reported in Table 8. Moreover, there was a dramatic decrease either in gait and in equilibrium control and in the combined synoptical measure (of total score) in the patients during follow-up (Table 9).
Results of Gait Tests in the Patients Observed During Follow-Up.

Results of Gait Tests at 60 Months: A Comparison Over Baseline, 12, and 24 Months.
Spearman’s rank correlation analyses indicated that there was a significant correlation between gait scores (total and separately, gait and equilibrium) and CDR scale scores (total Tinetti score/CDR: r = .81, P < .05 over 12 months; r = .83, P < .01 over 24 months; r = .89, P < .01 over 60 months; gait Tinetti score/CDR: r = .82, P < .01 over 12 months; r = .87, P < .01 over 24 months; r = .89, P < .01 over 60 months; and equilibrium Tinetti score/CDR: r = .81, P < .05 over 12 months; r = .83, P < .01 over 24 months; r = .89, P < .01 over 60 months).
Spearman’s rank correlation analyses indicated that there was a significant correlation between Tinetti total and equilibrium and gait score and BI over 12, 24, and 60 months (total Tinetti score/BI: r = .81, P < .05 over 12 months; r = .83, P < .01 over 24 months; r = .89, P < .01 over 60 months; gait Tinetti score/BI: r = .82, P < .01 over 12 months; r = .87, P < .01 over 24 months; r = .89, P < .01 over 60 months; and equilibrium Tinetti score/BI: r = .84, P < .01 over 12 months; r = .85, P < .01 over 24 months; r = .89, P < .01 over 60 months).
Furthermore, we have found a correlation between Tinetti equilibrium score and NPI over 24, 36, and 60 months (equilibrium Tinetti/NPI: r = .78, P < .05; r = .81, P < .01; r = .87, P < .01), Tinetti gait score and NPI over 24 and 60 months (gait Tinetti/NPI: r = .78, P < .05; r = .86, P < .01), and, of course, Tinetti total score and NPI over 60 months (Tinetti total score/NPI: r = .93, P < .01).
Spearman’s rank correlation analyses indicated that there was a significant correlation between gait scores (total scores and separately, gait and equilibrium) and AES-C (total Tinetti score/AES-C: r = .81, P < .05 over 12 months; r = .82, P < .01 over 24 months; r = .87, P < .01 over 60 months; gait Tinetti score/AES-C: r = .85, P < .01 over 12 months; r = .87, P < .01 over 24 months; r = .89, P < .01 over 60 months; and equilibrium Tinetti score/AES-C: r = .79, P < .05 over 12 months; r = .81, P < .01 over 24 months; r = .87, P < .01 over 60 months).
Surprisingly, we have found only a correlation between benzodiazepines intake and Tinetti equilibrium score at 12, 24, and 60 months (respectively, r = .77, P < .05; r = .78, P < .05; r = .67, P < .05 at 12 months; r = .81, P < .01; r = .77, P < .05; r = .84, P < .01 at 24 months; and r = .81, P < .01; r = .82, P < .01; r = .89, P < .01 at 60 months).
Considering neuroleptics, one should take into account the employment of typical or atypical neuroleptics. Moreover, quetiapine as an atypical neuroleptic shows less dopamine affinity than olanzapine, which we have examined through the study. Therefore, evaluating separately the results, we have observed the following relationships: Typical neuroleptics: A significant correlation was observed between haloperidol intake and Tinetti equilibrium score at baseline and at 12, 24, and 60 months (respectively: r = .61, P < .05; r = .72, P < .05; and r = .81, P < .01) and between haloperidol intake and Tinetti total score at 24 and 60 months (respectively: r = .81, P < .01 and r = .86, P < .01). No significant correlation was observed between promazine chloridate intake and Tinetti subscores at baseline and at 12 months. We have found a positive correlation between the equilibrium score of Tinetti test and promazine intake at 24 and 60 months (r = .74, P < .05 and r = .81, P < .01) and a positive correlation between the equilibrium score and the gait score of Tinetti test and promazine intake at 60 months (respectively: r = .79, P < .01 and r = .81, P < .01). Atypical neuroleptics: We have found a significant correlation between olanzapine intake and Tinetti equilibrium score at 24 and 60 months (respectively: r = .72, P < .05 and r = .74, P < .05) and between olanzapine intake and Tinetti total score at 60 months (r = .71, P < .05). We have found a positive correlation between the equilibrium score of Tinetti test and quetiapine intake at 24 and 60 months (respectively: r = .79, P < .05 and r = .82, P < .01). The mean dose of olanzapine remained stable during the 12th month up to the 60th month of follow-up (5.1-5.9 mg/d); on the contrary, quetiapine dosage increased up to 60th month of follow-up (56.9-89.9 mg/d).
Discussion
Subcortical VAD is a complex entity in which memory impairment is not so relevant as dysexecutive functions. However, it is still debated as a clinical nososgraphic unit. This work helps to consider 2 different specificities, which could help to distinguish sVAD from other dementia cluster syndromes, apathy and gait disorders.
Walking is a complex mechanism based on motor control, step rhythm, muscular activation and deactivation, motor adjustment, attention, perception, and so on. Spinal and brain stem activation, which seem to be fundamental for quadrupeds, in humans depend more on cortical and subcortical inputs. 37
The motor cortex contains several distinct areas in the frontal lobes, which receive inputs from sensory pathways, motor control structures, and modulatory pathways including the thalamus and BG (BG). This cluster of architectonically distinct frontal fields is fundamentally involved in movement planning and performance. 38 In the case of normal walking—an internally cued, well-learned motor act—it has been suggested that the supplementary motor area (SMA) engages in significant firing just prior to gait ignition. This probably reflects preparatory activity for each subcomponent of a movement sequence. 39 This preparatory activity may represent submovement program selection (ie, a complex set of instructions for each submovement), which is subsequently sent to the primary motor cortex (M1). It is thought that this SMA activity is switched off by phasic activity generated by the BG, which probably provides a nonspecific cue to trigger the submovement and to instruct the SMA to prepare for the next one. 9,40
The BG may thus provide the timing cues for switching between submovements in an “automatic” movement sequence and also cues new preparatory activity in the SMA. This interaction between phasic activity from the BG and premotor activity in the SMA is responsible for the smooth running of predictable, well-learned, and automatic movement sequences, which depend on internal cues. 9
As well pointed out by Rosano et al, the BG exert a compensatory mechanism in gait control, in particular in gait variability. In their study, subclinical brain vascular abnormalities were measured on brain MRI as infarcts and white matter hyperintensities. They demonstrated that greater variability in step length was associated with greater prevalence of infarcts and greater white matter hyperintensities severity, independent of age, gender, and cognitive function. 41 To implement these data, another study by Carboncini et al demonstrated that higher level gait disorders (HLGDs) are a common alteration in older people, and that gait disorders can occur in a neurological syndrome named as leukoaraiosis. 37 Data have been clinically confirmed by other works by the LADIS group, 13 by Srikanth et al, 42 and by Masdeu and Wolfson. 43
Liston et al hypothesize that an analogous situation may exist in HLGDs caused by microvascular alteration occurring in the SMA or its connections in the periventricular white matter or indeed infarctions in the BG. 9 This is in keeping with Meyer’s original concept of gait apraxia being caused by any mesial frontal lesion which is the anatomical site of the SMA. 44
It is also supported by current evidence suggesting that damage (periventricular white matter lesions or leukoaraiosis) to critical pathways, linking the BG to the ventrolateral nucleus of the thalamus and to the SMA and frontal areas, leads to abnormal gait lines in early VaD. 45,46 These critically placed infarcts presumably disrupt the timing cues from the BG in a similar way to PD and may therefore be expected to cause similar gait abnormalities. Well-accepted studies suggest and confirm that leukoaraiosis is associated with gait impairment, worse equilibrium scores, and falls. 3 -8,10 Some authors argue that these gait disorders may exist in mild subclinical forms associated with mild periventricular changes. Clearly, a severe gait disorder occurs in Binswanger’s disease in association with a severe leukoaraiosis. 9 However, a significant number of patients with vascular HLGDs do not have movement ignition and timing problems, related to the above-described pathological conditions but present with disequilibrium as their primary complaint. 6,9,47 Liston et al suggested that the primary disorder might be an infarction of the sensory/premotor area pathways and while the patients have no difficulties with automatic internally cued movements and as such the BG/SMA pathways are intact, they are unable to fully utilize sensory information from the environment, including proprioceptive, auditory, vestibular, and visual information to help initiate and control submovements. Such patients are unbalanced and unable to integrate external information into their movement sequences. Therefore, Liston et al distinguished 3 types of gait disorders in this patients, ignition apraxia, equilibrium apraxia, and mixed gait apraxia. 9
On the other hand, the evolution of subcortical dementia leads to magnification behavioral alteration as well as cognitive impairment worsening (in particular of executive function and frontal focusing). Apathy is a general tract of this group patients. We have observed it, purposely studied, and followed up. As a general observation, 48 the occurrence of apathy is connected to damage of prefrontal cortex (PFC) and BG 20 ; “emotional affective” apathy may be related to the orbitomedial PFC and ventral striatum; “cognitive apathy” may be associated with dysfunction of lateral PFC and dorsal caudate nuclei; and deficit of “autoactivation” may be due to bilateral lesions of the internal portion of globus pallidus, bilateral paramedian thalamic lesions, or the dorsomedial portion of PFC. 49 Previous studies have reported that in behavioral variant of frontal dementia (bvFTD), apathy is associated with changes in orbitofrontal cortex (OFC), 50,51 which has been postulated to be the anatomical correlate of “affective” apathy, 52 and with volume loss in the dorsal anterior cingulate and dorsolateral PFC. 53 Thus, it is possible that the AD apathy, described by Quaranta et al, 48 may reflect an alteration in OFC and its connections with subcortical nuclei (ventral striatum) that could be specific of bvFTD. Similarly, affective apathy may be related to an impairment of the so-called prosocial sentiments (such as guilt, pity, and embarrassment) connected to lack of empathy. Moll et al reported reduced social sentiments in patients with bvFTD; this deficit was related to hypometabolism in medial frontal polar cortex and septal area. 54
On the other hand, in AD, apathy severity has been connected to neurofibrillary tangles’ density in the anterior cingulate gyrus (ACG) 55 and to gray matter atrophy in the anterior cingulate and in the left medial frontal cortex. 55 -57 These findings were confirmed by a PET study showing the association of apathy with hypometabolism in the bilateral ACG and medial OFC. 58 -62 Additional regions of atrophy in the superior frontal gyrus, specifically BA 9, are also reported 56,63 as is atrophy of frontopolar (BA 10) and ventrolateral prefrontal regions, including BA 45 (pars triangularis). 63
One pathophysiological model for apathy in AD which addresses both structural and biochemical disruption is that of Guimarães et al. Their model sees ACG and OFC as part of a broader frontostriatal circuit, which is involved in decision making. Specifically, these regions are involved in evaluating action and outcomes and, via the basolateral amygdala and nucleus accumbens, feed into an ascending frontostriatal pathway to the dorsolateral PFC, which is ultimately responsible for selecting and executing behavioral responses. Damage to the ACG and OFC leads to a disruption of this circuit, resulting in impaired decision making and impaired response initiation, which presents as apathy. 64
This model resembles quite well our idea of apathy in sVAD. There is good evidence of high levels of apathy subcortical disease, such as PD, resulting from dysfunction at the striatal level, 65 and our data suggest the locus of dysfunction is at the cortical level, namely the ACG and the OFC, than in AD.
In sVAD, apathy might be the result of a wider prefrontal disease process or may suggest a putative role for these regions in mediating apathy, namely due to an involvement of the pars triangularis, of the superior frontal gyrus, and of the orbital operculum and may suggest that degeneration of the OFC is also implicated. Ventrolateral and superior frontal regions are also involved in the selection and execution of willed action and so may contribute to the diminished behavioral responses to everyday challenges displayed by apathetic patients. Recently, increased incidence of white matter hyperintensities in the frontal lobe has been associated with apathy 66 ; however, some studies have found no evidence of frontal involvement in apathy. 67 A recent and well-conducted study examined the relationship between behavior alterations and subcortical lesions (white-matter lesions and lacunes) in AD. Lacunes in the BG resulted in a 2- to 3-fold increased risk of delusions, hallucinations, and depression, when adjusting for cognition and atrophy. This suggests that BG lesions can contribute to the behavioral and psychological symptoms of dementia in patients with AD, independent of the AD process. 68
The results of our study might be considered at first instance as not unexpected and not completely novel. Although trying to evaluate them step-by-step, we could consider that they are the first results on gait and equilibrium in a selected population with sVaD , with a 60-month follow-up. Our prospective evidence demonstrates that in a selected group with sVaD, diagnosed accomplishing neurological and neuroimaging criteria, there are major evidence of significant gait and equilibrium problems, from the beginning of the observational period and their worsening during the 3 years of follow-up. A clear point should be made standing from the firm exclusion criteria of brain stem lesions, in order to eliminate potential confounding effects of specific ischemic lesions on gait and equilibrium. Therefore, gait and balance alterations observed in this population should be considered as a result of subcortical widespread damage and not related to other problems, such as pharmacological effects or superimposing comorbidites. During follow-up, the gait abnormalities are stressed by the general worsening, the decrease in behavioral control, and the consequent neuroleptic and benzodiazepine intake. At the same time, apathy is a primary consequence of sVAD, which increments with the prolongation of clinical observation but is present form the first visit of these patients.
Therefore, it might be concluded that subcortical lesions, per se, cause imbalance and gait alteration and apathy; we might hypothesize that these subcortical hypoperfusion might interrupt long loop reflexes of deep white motor tracts and descending motor fibers arising from medial cortical areas. 12,69,70 Moreover, subcortical vascular lesions involve fibers connecting frontal cortex and subcortical structures, which are responsible for motivation, executive function, planning, and attention too (see in particular frontal eye fields). It has been suggested 69,70 that the BG maintain cortically selected motor set in the SMA and provide internal cues to the SMA in order to enable each submovement to be correctly linked together. 71 It has been demonstrated that attention problems, in particular attentional shift and focused attention, decrease the performance of gait in parkinsonian patients with freezing of gait. 72 -74
These data seem to be in accordance with what we observed at baseline: subcortical demented patients might show gait problems just for their vascular altered white substance.
Therefore, all these considering, we suggest the following different conclusions: Subcortical VAD should be considered a single and well-determined reality. Subcortical VAD has 3 main peculiarities, frontal impairment, as dysexecutive alterations and not memory impairment, gait and equilibrium alterations, and apathy. Patients with sVAD interface with other major clinical events, and their pharmacological treatment could worsen the evolution of the neurological events.
Considering all these facts, sVaD may be associated with gait and balance alteration per se; its worsening and the pharmacological choices, imposed by the evolution of the neurological condition and comorbidities, should be considered by the clinicians in order to prevent major gait alteration, falls, and postural imbalance or at least to reduce their consequence in a real frail population.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.