
Abstract
Loss of synaptic function is critical in the pathogenesis of Alzheimer’s disease (AD) and other central nervous system (CNS) degenerations. A promising candidate in the regulation of synaptic function is Shank, a protein that serves as a scaffold for excitatory synaptic receptors and proteins. Loss of Shank alters structure and function of the postsynaptic density (PSD). Shank proteins are associated with N-methyl-
Introduction
In Alzheimer’s disease (AD), recent attention has been drawn to postsynaptic neuronal structures as the anatomic site of dysfunction leading to cognitive symptoms and signs. Shank proteins are a family of multidomain proteins located beneath the postsynaptic membrane. 1,2 They form the postsynaptic platform that resides within the postsynaptic density (PSD). 3 Isolated independently by a handful of investigtors in 1998 and 1999, shank proteins are the product of 3 genes—Shank1, Shank2, and Shank3.
Shank plays a critical role in integrating the various postsynaptic membrane proteins, including the different types of glutamate receptors: N-methyl-
Shank proteins and associated glutamate receptors participate in a concerted manner to form spines and functional synapses. 7 In addition to their roles in synaptic function and the structure of dendritic spines, Shank proteins have gained attention over the past decade for their roles in autism spectrum disorders (ASDs) 8 and Schizophrenia. 9 More recently, Shank has been implicated in another cognitive and behavioral pathology, AD. 10,11 These findings indicate the posible common pathogenic role of Shank protein in cognitive-deficient neurological diseases.
Shank Protein Family and Distribution
The family of Shank proteins is comprised of 3 members 12 —Shank 1, Shank 2, and Shank3—that differ from each other through alternative splicing of messenger RNA (mRNA). 4 Due to simultaneous investigation by several groups 13 –18 and its distribution in various tissues, Shank proteins (named for containing SH3 domain and ankyrin repeats) have several alternative names, including proline-rich synapse-associated protein (ProSAP), 1,12 cortactin-binding protein (CortBP), 15 Spank, Somatostatin receptor-interacting protein, 19 and Synamon. 2,17
Members of the Shank family share a common structure of multiunit domains consisting of multiple ankyrin repeats, SH3, PDZ, a proline-rich domain, and a sterile α motif (SAM). 20 Although these domains may also be found in other protein families, they share a greater degree of amino acid sequence and structural similarity than proteins belonging to other families. For example, Lim et al found 63% to 74% identical amino acid sequence in the SH3 domain of Shank1, Shank2, and Shank3. 21 When compared with the SH3 domain of the Src protein, only 20% to 24% matched the SH3 domain of the Shank family. This was likewise observed in the PDZ domains of Shank1, Shank2, and Shank3, which shared 82% to 88% similarity with one another, whereas the PDZ domain within PSD-95 only shared 24% to 25% similarity. 21
Shank’s domains can assemble into large sheets within the PSD or interact with synaptic or cytoskeletal proteins within dendritic spines and PSDs. 3,4,8,14 Shank thereby acts as an essential scaffold that provides structural support for several proteins at once. Isoforms of Shank1, Shank2, and Shank3 are formed by alternative splicing of Shank mRNA 21 in particular distributions of tissues or when transcription and translation have different starting sites. 4,21 Sites of alternative splicing were first identified by mismatched sequences of amino acids when paired with complementary DNA clones and then confirmed by analysis of real-time polymerase chain reaction (RT-PCR); the distribution of Shank mRNA was identified by Northern blot.
Shank proteins have been localized in several tissues. Shank1 is found in the brain, 17,20 –22 cochlea, 23 and retina 24 ; Shank2 is in the bran, liver, and kidney 15,21 ; and Shank 3 is in the brain, heart, and spleen. 21 All ProSAP/Shank family members are expressed in the brain. Shank1 transcripts are found in the cortex, hippocampus, purkinje cell layer of the cerebellum, thalamic nuclei, hypothalamic nuclei, msesncephalic nuclei, and amygdaloid nuclei. Shank2 transcripts are found in the cortex, hippocampus, purkinje cell layer of the cerebellum, caudate, putamen, thalamic nuclei, mesencephalic nuclei, and amygdaloid nuclei. Shank3 transcripts are found in the cortex, hippocampus, granular cell layer of the cerebellum, caudate, putamen, thalamic nuclei, hypothalamic nuclei, and amygdaloid nuclei. Labeling of Shank mRNA is exclusive to the dendrites 25 at these locations, while Shank proteins are present at the PSD of excitatory synapses. 1,14 The expression and distribution of Shank proteins in the brain and other organs are listed in Table 1. Shank1 protein is found throughout the dendritic spine, Shank2 protein is found preferentially concentrated at the PSD. 26 Varied recruitment of the isoforms within tissues and within neurons suggest variation in their function. 25
Expression Pattern of Shank Transcripts. 25
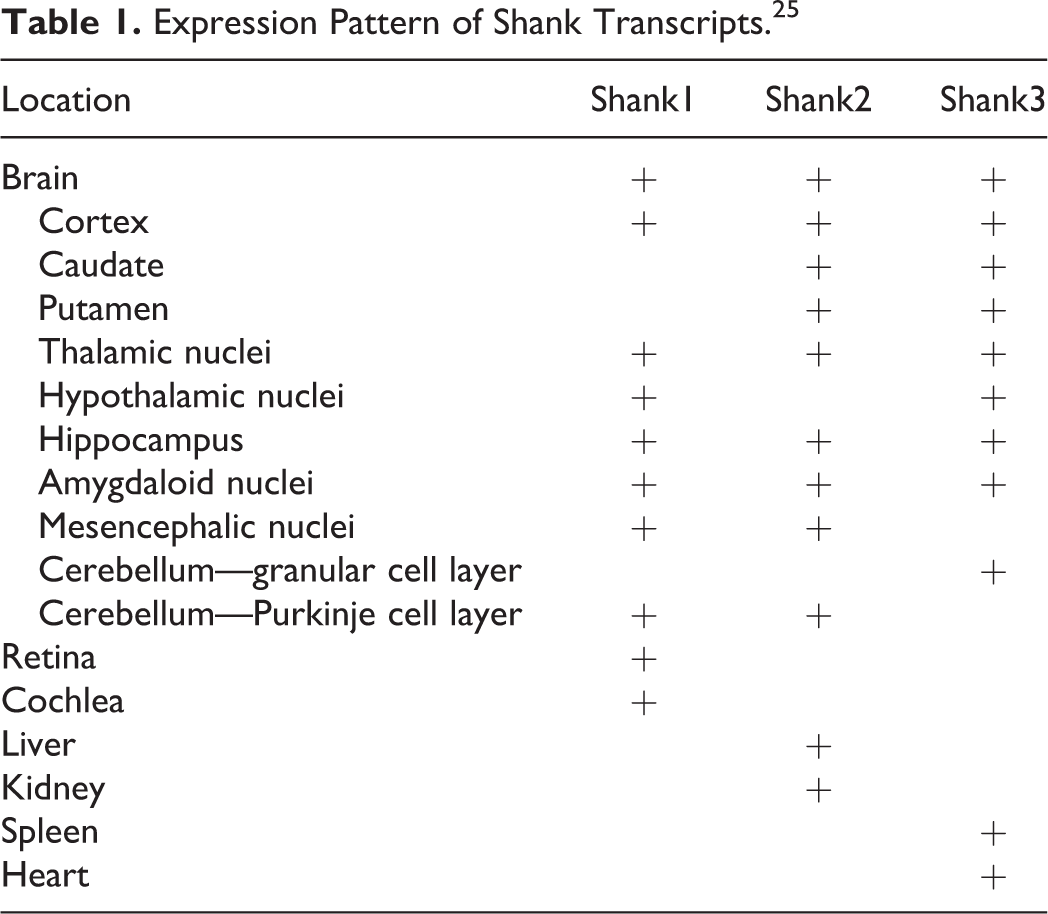
Alternative splicing occurs not only within different tissues but also during different stages of development. For example, Lim et al demonstrated via RT-PCR analysis that the 2 variations of Shank1 that resulted from splicing at site 1 were invariably and stably expressed in cortex and cerebellum of developing and adult rat brains. However, 1 of the 2 variants of Shank1 that underwent splicing at site 2 was upregulated significantly in the developing cortex, while the other variant was downregulated; this variation in expression was not observed in the developing cerebellum. 21
Differential expression of Shank genes may explain preferential and sequential loss of particular subsets of neurons in AD brains. Extensive probing by Boeckers et al demonstrated localization of Shank mRNA transcripts in different stages of development of rat brain. 12 Although all Shank transcripts are found in cortex and hippocampus, variation occurs in other brain structures. Transcripts of Shank1 and Shank2 are slightly higher at birth than in development. This is unlike Shank3 that begins increasing with age, approximately 16 days after birth. Shank2 is one of the primary proteins incorporated into the structure of synapses—even before SAP90/PSD-95 and NMDA receptor subunits are present. 12 This implicates Shank as an essential scaffolding protein.
Shank Protein Network in the PSD
The Shank proteins are considered the “master organizing” molecules of the PSD 3,14 due to their roles in forming large sheets and interacting with several protein complexes and cytoskeletal elements. Each of Shank’s 5 domains makes unique interactions that provide scaffolding for the PSD. The NMDA receptors are anchored by PSD-95/SAP90 (PSD-95/guanylate kinase-associated protein [GKAP]), 3 which in turn interacts with the Shank PDZ domain. Important modular protein interaction domains that are essential for the organization of the PSD involve PDZ (PSD-95, DLG, ZO1). The mGluR is tightly linked to the NMDA/SAP90/PSD-95 complex, and AMPAR GluR1 interacts directly with the PDZ domain of ProSAP/Shank. In a similar manner, AMPARs are anchored by glutamate receptor AMPAR binding protein (ABP)-interacting protein that interacts directly with the Shank PDZ domain. 27 The mGluRs are anchored by Homer/Vesl and Shank may bridge the Homer-mGluR receptor complex and the PSD-95/GKAP-NMDA receptor complex, 21 as Homer interacts with the proline-rich domain of Shank. 3 Homer may also interact with calcium and potassium channels. 28,29 This intricate web of connections is further strengthened by Shank’s association with itself through interactions at the SAM domain. Finally, the platform created by Shank attaches to F-actin cytoskeleton and smooth endoplasmic reticulum. 3 Extensive scaffolding by Shank organizes a wide range of membrane proteins and receptors. Because of its direct interaction with NMDA and other glutamate receptors, Shank proteins are recoginized as organizer in PSD to maintain synaptic function (Figure 1).
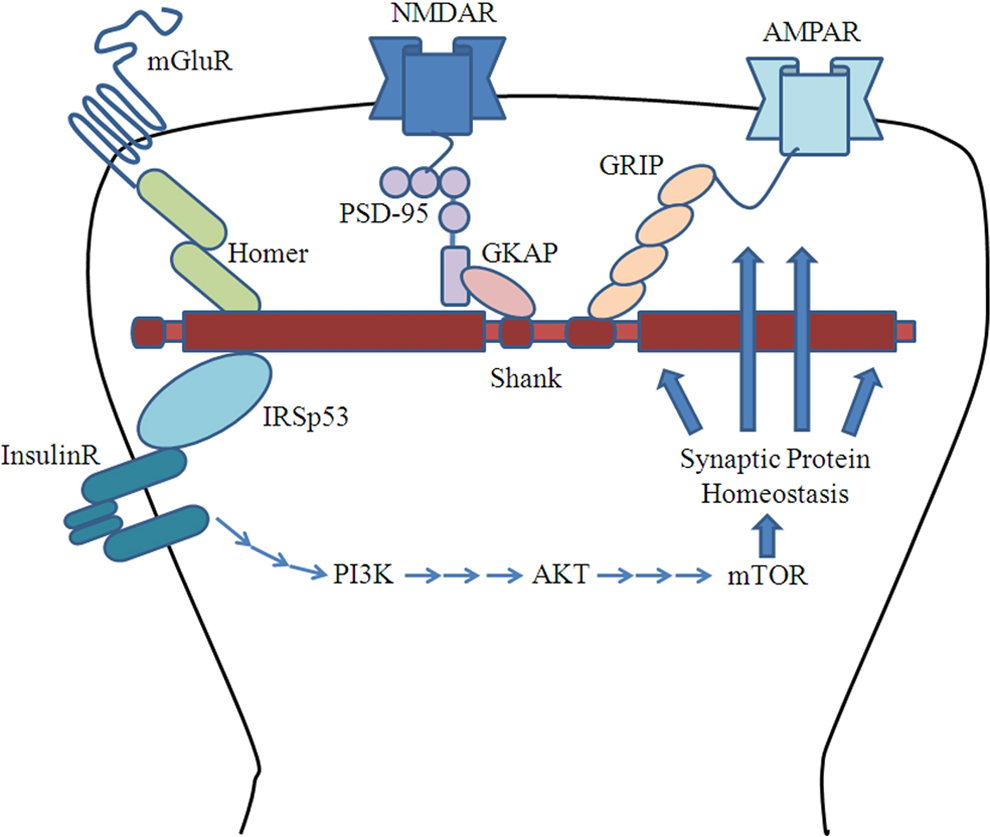
Shank-protein network in postsynaptic density (PSD).
Shank Protein Homeostasis at the Synapse
Shank protein is synthesized at dendritic spines through regulation by mammalian target of rapamycin (mTOR) 30 –32 and is degraded by the ubiquitin proteasome system. The complex formation of PSD95-GKAP-Shank induces synaptic formation. 33 In hippocampal neurons, overexpression of Shank1 causes morphological change in dendritic spines. 25,34 Sala et al go on to claim that Shank1 concentrations correlate with synaptic strength; increased Shank1 leads to increased strength of the synapse. A similar correlation is made between Shank2 and purkinje cell synaptogenesis and dendrite structure, as more Shank2 mRNA is present when synaptogenesis is most active. 25 Additionally, depolarization of neurons causes Shank relocation to the PSD; Shank1 has dynamic movment while Shank2 is relatively more stable. 26 Interestingly, the hormone insulin is highly effective in inducing AMPAR recycling at PSDs in hippocampal culture experiments. 35 This may contribute to the role of insulin in improving long-term potentiation and memory, which will be discussed further (Figure 1).
Shank Protein Mutation in Mice
Mice with different Shank protein mutations and their effects on behavioral alterations (eg, cognitive function) and synaptic changes are listed in Table 2. This suggests correlations that are observed in humans.
Shank Mutations in Mice and Associated Behavioral and Synaptic Changes.
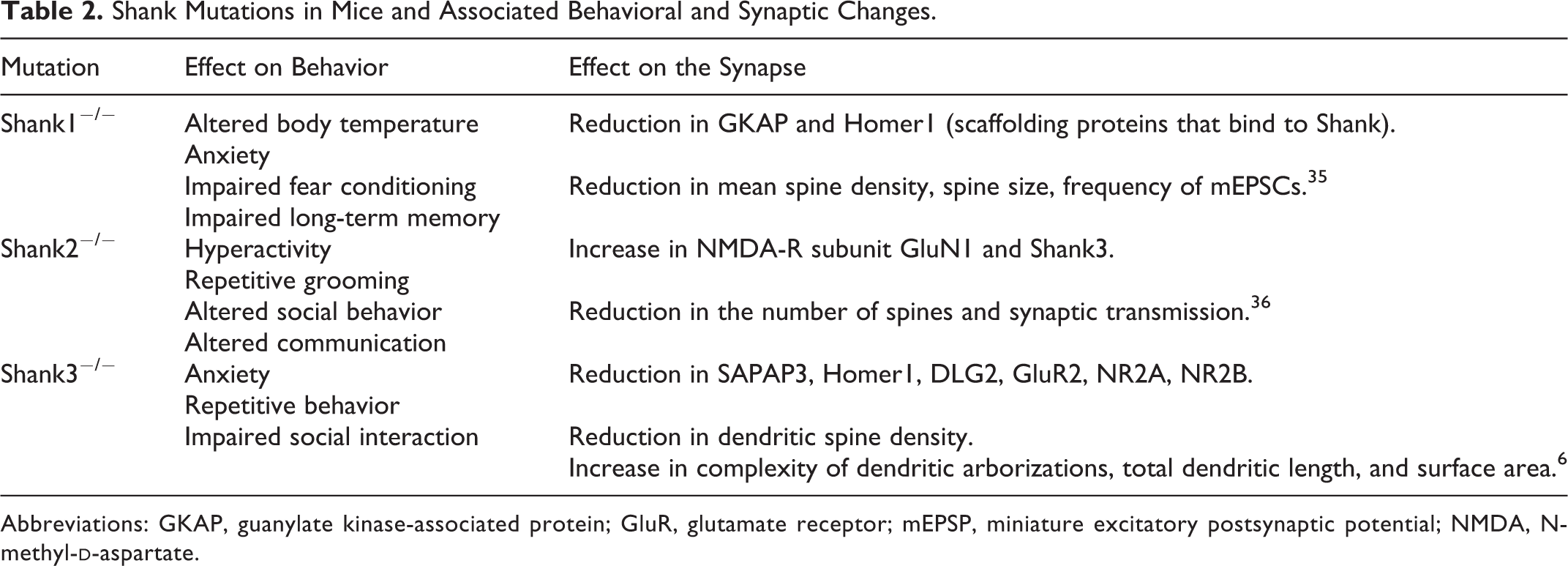
Abbreviations: GKAP, guanylate kinase-associated protein; GluR, glutamate receptor; mEPSP, miniature excitatory postsynaptic potential; NMDA, N-methyl-
Shank1 Mutation
Null mutation Shank1 mice develop smaller postsynaptic densities, smaller dendritic spines, 22,35 and decreased spine density. 35 Physically, no obvious abnormalities are seen in Shank1 knockout mice; this includes evaluation of general health, empty cage behaviors, and reflexes. The only statistically significant difference between Shank1−/− and control mice is in body temperature. Behaviorally, however, the null mutation mice exhibit higher anxiety, 35 reduced “open field activity,” and altered learning and memory 22 exhibited by impaired fear conditioning. 35 Spatial memory is enhanced but long-term retention of the memory is impaired. 35 Shank1−/− mice do not display any difference in cumulative self-grooming time when compared with Shank1+/+ and Shank1+/− mice. However, Shank1−/− mice do exhibit mild anxiety-like phenotype as measured by reduced number of transitions between light and dark compartments. Yet this may be confounded by reduced neuromuscular strength of Shank−/− mice, which is based on latency to falling from a suspended wire, latency to falling from a Rota rod, and distance traversed. Overall, the authors concluded that Shank1 null mutation affects cognitive ability, fear, and memory, rather than sociability. 22 However, other groups have demonstrated that Shank1 null mutant mice display impaired communication. 36
Shank 1 has been implicated in learning and memory dysfunction of Aβ(1-42)-injected AD model rats. 37 The Aβ disrupts scaffolding proteins PSD-95, Homer1b as well as Shank1, resulting in declustering and thinning of the PSD. This protein turnover is reportedly independent of proteasome activity, as pretreatment with proteasome inhibitor does not protect Homer1b or Shank1. 38
Shank2 Mutation
Shank2−/− mutants exhibit behavioral changes including hyperactivity, repetitive grooming, and alterations in social behavior and communication. 39 Variants of Shank2 have reduced spine volume and smaller cluster sizes. 40
Shank3 Mutation
Mutation in Shank3 leads to modified ubiquitination of Shank3 protein, which results in the loss of NMDA receptors within an aberrant PSD structure. 6,8,41 Shank3−/− mutant mice similarly display repetitive behavior and impaired social interaction representative of autism. Social interaction is impaired, as Shank3−/− mice prefer interacting with an empty cage rather than interacting with other mice. This contrasts with wild-type mice that spend more time in cages of novel mice than in empty cages. Additionally, repetitive anxiety-associated behavior such as grooming was continued to an extent that was self-injurious; lesions on the back of the neck and face progressed to large parts of the body. This possibly corticostriato dysfunctional behavior was not present in Shank 3+/− mice that were housed in the same cages as Shank3−/− mice. 6
In mice, Shank has been shown to regulate the structure, maturation, and organization of dendritic spines. Rat Shank mRNA shows elements that are conserved in humans. 25
Shank Protein Pathological Changes at Synapses in Neurological Diseases Affecting Cognition
Shank proteins are believed to be involved in a signaling pathway that is shared and disrupted in several neuropsychiatric disorders. 2
Autism Spectrum Disorders
Shank mutations have been shown to be involved in ASDs. Such mutations traced in families include nonsense mutations, frameshift mutations, 8,42 –45 and abnormal doses of genes, thus resulting in varying degrees of cognitive deficits, language/speech disorders, anxiety disorders, 45 and ASD. 8 Loss of Shank2 alleles and translocation in 11;17:19 46 are associated with severe behavioral anomalies, language impairment, and intellectual disability. 42,46 –48 Truncation of the C-terminal of Shank3 impairs proper mGluR and actin. 8,49,50 Translocation in 22q13.3 deletion syndrome disrupts Shank3, resulting in Phelan-McDermid syndrome, an ASD. 9 Several additional duplications and deletions in Shank3 have been targeted in ASD, 8,9 suggesting the role of Shank in neurodevelopment. 8,42,43,51,52
Schizophrenia
Likewise, the role of shank in schizophrenia has been a recent target of investigation. Although schizophrenia likely has a multifactorial etiology, many linkage studies have described the involvement of Shank de novo mutations in the development of schizophrenia. 9,52 Genetic linkage of schizophrenia within families has been correlated with the 22q11-13 locus 53 –57 that also codes for Shank3. Individuals afflicted with schizophrenia have linearly correlated cognitive impairment 9 and associated cognitive loss. 58 Lennertz et al studied the correlation between Shank1 variants and reduction in auditory working memory in patients with schizophrenia. It is commonly known that working memory declines in AD, 59 and in schizophrenia it has been suggested that working memory is the rate-limiting capacity. 60 This underlies the possibility of a common role of Shank in memory formation in both schizophrenia and AD. Interestingly, Critchlow et al have found that the atypical antipsychotic clozapine increases Shank1 density in rats. 61 This finding may be of therapeutic interest in the treatment of other Shank-related diseases affecting cognition.
Alzheimer’s Disease
Despite its association with ASDs and schizophrenia, few groups have shown the association with AD using human tissues. Gong et al published evidence that protein levels of Shank3 were significantly reduced in the PSD fractions of human AD brains. Abnormal polyubiquitination may be involved in Shank protein regulation in human AD. 10 Furthermore, the relation of glutamate receptors, neuronal excitotoxicity, and long-term potentiation in memory formation may be molecularly connected to Shank and its organization of the postsynaptic platform and glutamate receptors. Pham et al also detected a similar association between Shank1, Shank3, and AD-model mice brains. Levels of Shank1 and Shank3 were reduced in AD APP transgenic mice compared to controls. 11
Insulin in the Treatment of Shank-Relevant Central Nervous System Diseases Affecting Cognition
Insulin Receptor-mTOR Signal Pathways
Although the role of insulin receptors at neuronal synapses is not completely understood, 32 it has been suggested that insulin likely regulates gene expression 62 and translocation of GABA-A receptors to the synaptic plasma membrane. 63 Lee et al demonstrated that insulin selectively increases translation of PSD-95 protein in the CA1 region of the hippocampus. 32 This synthesis of PSD-95 is blocked by pretreatment with rapamycin, suggesting that the insulin pathway involves mTOR and PI3K. Furthermore, insulin induces increased phosphorylation of PI3K downstream effectors—PDK1, Akt, and mTOR—as well as other translation-related protein factors in dendritic spines, such as 4E-BP1 and p70S6K. 32 This implicates the role of insulin in the PI3K/Akt/mTOR pathway, which has been previously implicated in apoptosis and many cancers. 64 According to the pathway, PI3K phosphorylates lipids such as phosphatidylinositol-3,4,5-triphosphate that acts as a second messenger to recruit the serine/threonine-specific protein kinase Akt. The Akt in turn activates mTOR, which is another serine/threonine protein kinase that is able to regulate protein translation through phosphorylation of several intracellular effectors, one of which may be Shank. The possibility of an insulin/mTOR/Shank3 pathway needs further investigation (Figure 1).
By activating the insulin receptor, insulin also activates other accessory molecules such as IRS. 65,66 The IRSp53 is a component of the postsynaptic density that links to Shank1, Shank3, 67 and PSD-95 within the postsynaptic NMDA receptor complex. The proline-rich region of Shank associates with the SH3 domain of insulin receptor tyrosine kinase substrate IRSp53 67 and is regulated by cdc42. 68,69 The IRSp53 is found in the cortex, hippocampus, cerebellum, and caudate putamen—areas that historically have high expression of Shank—and IRSp53 additionally colocalizes and coprecipitates with Shank3. 67 Primary cultured neurons of IRSp53 knockout mice have impaired dendritic growth, decreased PSD size, and compensate with increased numbers of NMDA receptor subunits. 70 Knockout IRSp53 mice display impaired memory and have trouble remembering which cage delivered electrical foot shocks. Other behavioral abilities such as motor coordination and exploratory behavior are unaffected by gene mutation, suggesting the role of IRSp53 in long-term potentiation. 70 Insulin has been studied in several central nervous system (CNS) diseases, 71 and in particular, the clinical effects of intranasally administered insulin have been of recent interest. This route is used to minimize systemic distribution and peripheral effects of insulin but has mixed effects. Insulin receptor-mTOR signal pathways could be used to regulate the synaptic protein homeostasis in these CNS diseases.
Autism Spectrum Disorders
Stern draws similarities between the PI3K/Tor intracellular pathway of insulin signaling that matches the signaling pathway implicated in autism. 72 Although no direct clinical trials have been conducted on autism and insulin, there have been studies on insulin-like growth factor 1 (IGF-1) and pioglitazone. Pioglitazone is a thiazolidinedione, which modulates sensitivity to insulin. In a case study of 25 children with ASD who were given pioglitazone for 3 to 4 months, researchers noted an improvement in behavioral irritability, lethargy, stereotypy, and hyperactivity. 73 A clinical study evaluating IGF treatment in patients with Shank3-related ASD (Phelan-McDermid Syndrome) is currently underway. 74
Schizophrenia
The most recent research on insulin therapy in schizophrenics does not seem to show an effect on psychopathology nor cognition. 75,76 However, further investigation needs to be conducted.
Alzheimer’s Disease
Insulin signaling has been implicated in memory impairment and AD, as insulin has been shown to protect against pathogenic phosphorylation of tau 77 as well as binding of β-amyloid. 78 Blocking of mTOR activity has a similar effect on tau and Aβ misfolding, with additional restoration of cognitive function in animal models. 79 –81 Several clinical trials on intranasal administration of insulin report improvement in memory impairment 82 –85 and reduced risk of developing AD. 86,87 In addition to insulin, agents that facilitate insulin signaling and are Food and Drug Administration approved for diabetes have been studied in AD. Rosiglitazone may increase dendritic spine density to improve cognition in patients with AD. 88 Liraglutide prevents synapse loss, prevents deterioration of synaptic plasticity, and prohibits the formation of β-amyloid plaques in the cortex of AD brains. 89 Exendin 4 (exenatide) may improve behavioral measures of cognition in transgenic AD mice. 85 These studies suggest that insulin and insulin sensitizers could be used to improve cognition in patients with AD.
Conclusion
Synaptic dysfunction is gaining attention as a key cause of cognitive loss in chronic disease. Although both pre- and postsynaptic structures have been studied, many investigators agree that postsynaptic changes are contributory. Thousands of proteins are integrated in the PSD, and Shank proteins have been shown to be essential in protein organization, signaling pathways, and cytoskeletal elements at the PSD. The loss of shank proteins in neurological disorders with cognitive and behavioral decline, such as ASD, schizophrenia, and AD, suggests that pathological changes at the PSD may be the common pathway of these disease processes. Strategies to maintain Shank proteins may be explored as a novel target to maintain or stabilize behavior and memory in neurological diseases with cognitive dysfunction. Additionally, more evidence needs to be found to support the fact that insulin-mTOR signaling pathway may be used to normalize protein homeostasis at the Shank-postsynaptic platform, thereby improving synaptic function in the treatment of neurological diseases affecting cognition and behavior.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: NIH1R21AG031388 (YG) and Potamkin Foundation (CFL).