
Abstract
Early-onset dementia, presenting before age 65 years, is increasingly recognized. It is often difficult to diagnose, since non-Alzheimer’s etiologies and unusual dementias are common. These conditions are more commonly genetic, and important potentially inherited causes of early-onset dementia include early-onset Alzheimer’s disease, frontotemporal dementia, Kufs’ disease, and Niemann-Pick disease type C. For each of these diseases, this review provides information on common clinical presentations, etiology, pathophysiology, and current and experimental treatments. A discussion of the diagnosis and workup for early-onset dementia is included with an emphasis on conditions that may have other involved family members.
Keywords
Introduction
Dementia is a common disabling condition with a huge impact on social, financial, and public health. Alzheimer’s disease (AD) is the most common subtype, especially in late-onset dementia cases. In the United States, it is estimated that 5.4 million Americans have AD, and a new AD case is diagnosed every 70 seconds. 1 The diagnosis and management of dementia can be challenging when symptoms do not fit easily into an Alzheimer’s pattern. Diagnosis of early-onset dementia (EOD) is particularly problematic because AD comprises a smaller proportion of this group. There is a wider array of potential neurological and psychiatric causes in early onset disease, some of which may mimic each other. The EODs are more often inherited as either autosomal dominant or recessive conditions. The purpose of this review is to help guide physicians in the differential diagnosis of early-onset dementia with an eye toward familial conditions. The review includes a discussion of the differential diagnosis and workup, along with a diagnostic algorithm.
Early-Onset Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common cause of dementia and represents a major public health concern. 2 It has been estimated that in 2000, there were 4.5 million individuals in the United States over the age of 65 years with Alzheimer’s disease. 3 This number is expected to grow to 13.2 million in 2050. 3 The Alzheimer's disease occurring before the age of 65 years is less common and is referred to as early-onset Alzheimer’s disease (EOAD). A study in England population found the prevalence of AD to be 17.4 and 35.0 of 100 000 persons in individuals aged 30 to 64 and 45 to 64 years, respectively. 4 A more recent study in the same country found that in individuals aged 45 to 64 years, the prevalence of EOAD was 24.2 of 100 000 persons. 5
A number of genetic factors have been identified in the pathogenesis of AD, some of which cause EOAD. In a family with a history of EOAD confirmed by autopsy, Goate et al examined cosegregation of AD and markers on the long arm of chromosome 216 and found a point mutation in the amyloid precursor protein (APP) gene. 6 In a subsequent study, a novel gene called presenilin 1 (PS-1) was discovered on chromosome 14, along with 5 distinct missense mutations that cosegregated with EOAD. 7 Soon afterward, PS-2, a gene similar to PS-1 in nucleotide and amino acid sequence, was found on chromosome 1. 8,9 Three missense mutations in PS-2 were found in individuals affected with AD. 8,9 Another gene of interest that has been studied is apolipoprotein E (ApoE) and, more specifically, the ApoE4 allele. The ApoE4 allele has been associated with increased amyloid-β deposition 10 and, compared with ApoE2 and ApoE3, ApoE4 is overrepresented in patients with late-onset AD. 11 Individuals with 2 copies of the ApoE4 allele may have early-onset inheritance. While mutations in APP, PS-1, and PS-2 are autosomal dominant, many cases of EOAD are autosomal recessive. 12 From a study of concordance among parents and offspring, it was concluded that EOAD is almost entirely genetic, with over 90% of cases being autosomal recessive and less than 10% resulting from dominant mutations. 12 Currently, genetic testing is available for the autosomal dominant forms of EOAD. Athena Diagnostics, Inc (Worcester, Massachusetts) offers an EOAD evaluation that includes ApoE testing, an APP DNA sequencing/duplication test, and DNA sequencing tests for PS-1 and PS-2.
The prevailing hypothesis explaining the pathophysiology of Alzheimer’s disease is the amyloid hypothesis. The APP is cleaved by β-APP cleaving enzyme and γ-secretase, and the amyloid-β peptide is excised as a result. 13 According to the amyloid hypothesis, amyloid-β peptide is deposited in brain parenchyma and causes neuronal dysfunction and death, ultimately leading to dementia in AD. 13 Deposition of amyloid-β peptide leads to the formation of plaques, one of the histological hallmarks of AD. The microtubule-associated protein tau then becomes hyperphosphorylated, forming the key pathological neurofibrillary tangle (NFT). 14 Both plaques and NFTs can be visualized with thioflavin S or silver impregnation technique (Figure 1). It has been noted that tau pathology correlates with the severity of dementia in AD. 15
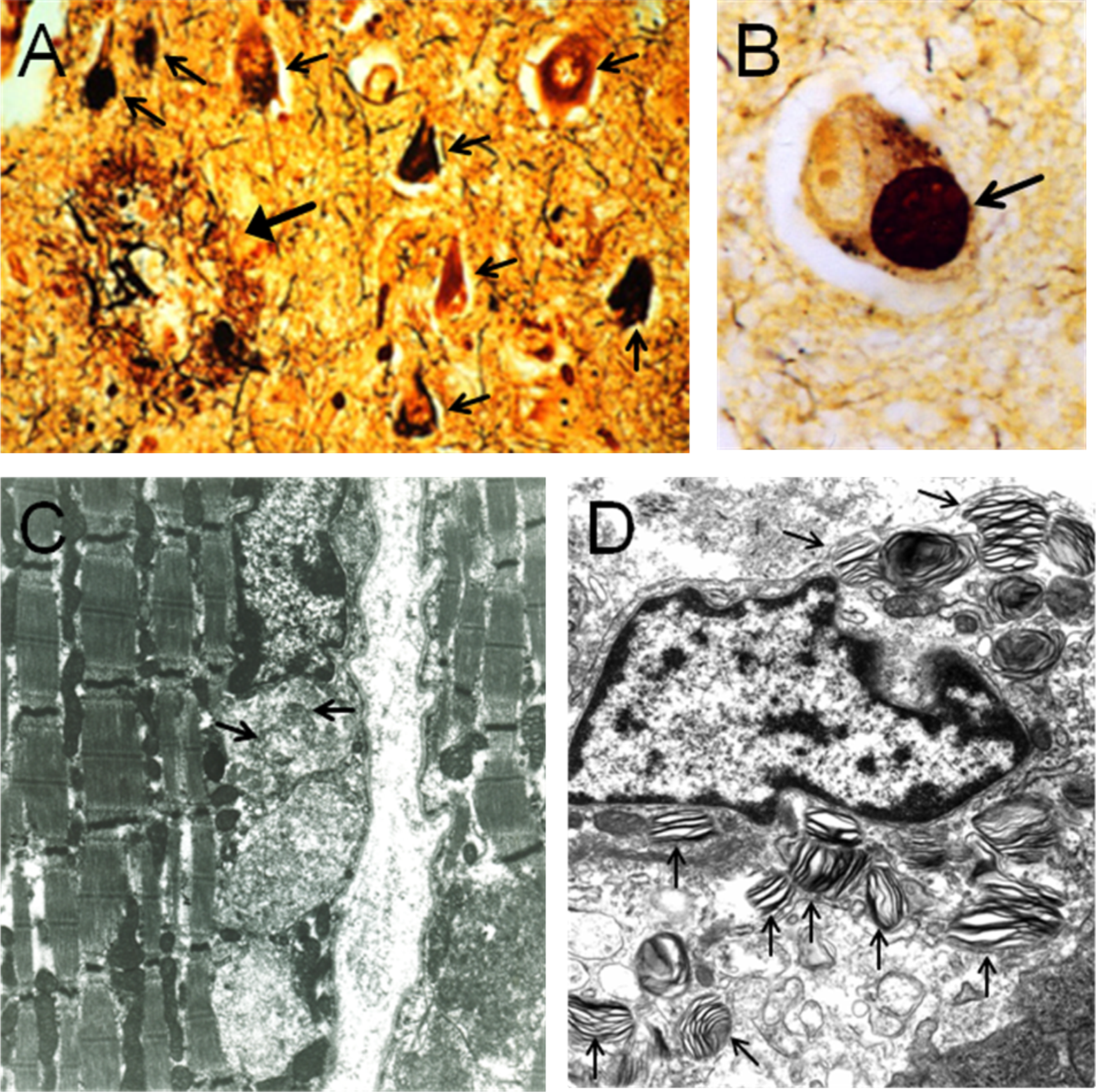
Histological findings in genetic causes of early-onset dementia. A, Bielschowski stain at ×200 of brain specimen of patient with Alzheimer’s disease. A plaque (large arrow) and multiple neurofibrillary tangles (smaller arrows) are visible. B, View of brain specimen of patient with frontotemporal dementia (×600). Pick body (arrow) is present. C, Deposits in the muscle of a patient with neuronal ceroid lipofuscinosis (arrows) are visualized with electron microscopy. D, Electron micrograph of tonsillar macrophage (foam cell) containing zebra bodies (arrows) in a patient with Niemann-Pick disease. Electron micrographs courtesy of Dr Jean-Pierre de Chadarevian, Department of Pathology and Laboratory Medicine, Drexel University College of Medicine, St. Christopher's Hospital for Children, Philadelphia, PA.
Patients with AD experience episodic memory impairment that worsens and progresses to other cognitive domains over time. In cases of EOAD, patients present more frequently with atypical clinical manifestations such as visual impairment, executive dysfunction, behavioral changes, or language difficulties. 16 In a recent study, 62.5% of patients with EOAD presented initially with typical episodic memory dysfunction, and 32% of this group had depression at onset. 17 The remaining 37.5% of patients had an atypical presentation, the most common of which was behavioral and executive dysfunction. 17 Noncognitive symptoms including seizures, myoclonus, and Parkinsonism are also common in EOAD. 18
Current Food and Drug Administration-approved therapies for AD include cholinesterase inhibitors and the N-methyl-
While current therapies for AD do not impact the progression of the dementia, experimental approaches are aimed at developing disease-modifying treatments. Low molecular mass compounds, such as methylene blue, appear useful in preventing tau aggregation and the formation of fibrils. 22 Impaired axonal transport and microtubule function are found in tauopathies, and microtubule-stabilizing drugs have been used with some success. 23 Drugs that can assist in the folding of hyperphosphorylated tau and prevent tau aggregation, such as FKBP52, a co-chaperone of HSP90, may provide benefit. 23 A variety of kinases and kinase inhibitors have also been used. 23 Additionally, studies in mice have shown that amyloid-β-directed vaccination can prevent the development of plaques and behavioral symptoms. 23 A number of clinical trials involving antibody fragments and humanized amyloid-β-specific antibodies are underway. 23 Other experimental approaches involve α-secretase activators, β-secretase inhibitors, γ-secretase inhibitors, and amyloid-β aggregation inhibitors. 24
Frontotemporal Dementia
Frontotemporal dementia (FTD) is a leading cause of dementia, particularly in patients less than 65 years of age. 25 In a province of the Netherlands, prevalence was estimated to be 4 to 9 of 100 000 persons. 26 In Cambridgeshire, UK, the incidence of FTD in the age range of 45 to 64 years was 3.5 per 100 000 person-years. 27 Another study from 1990 to 1994 in Rochester, MN, showed that the incidence rates per 100 000 person-years were 2.2 for the age of 40 to 49 years, 3.3 for the age of 50 to 59 years, and 8.9 for the age of 60 to 69 years. 28
There appears to be a strong genetic component to the development of FTD. Analysis of pedigrees in one study showed that 37.6% of patients had significant family history. 29 More than 100 families with mutations of the MAPT gene on chromosome 17 have been found. 30 The clinical entity that results from these mutations is called FTD and Parkinsonism linked to chromosome 17 or FTDP-17. 30 Pathologic tau and tau inclusions are present in FTDP-17, which is classified as a tauopathy. 30 Mutations in PGRN, the gene that codes for progranulin, result in FTD with ubiquitin-positive inclusions (FTD-U). 31,32 It has been found that the decreased expression of PGRN results in pathological processing of TAR DNA-binding protein-43 (TDP-43) by caspase 3. 33 In some cases of FTD, TDP-43 becomes ubiquitinated and accumulates in neurons. 25 In such cases, ubiquitin-positive and TDP-43-positive inclusions may be visualized on histological analysis. The role of TDP-43 in the pathophysiology of FTD is unclear. 25
Additional changes found in the FTD brain include superficial cortical spongiosis and gray and white matter gliosis. If Pick cells (α-B-crystalline positive neurons) and Pick bodies (tau-positive round neuronal inclusions) are present, the disease is referred to as classical Pick’s disease (Figure 1). On gross anatomical examination of the brain, there is a “knife-edge” atrophy of the frontal and anterior temporal lobes. 30
Patients with FTD are classified into 3 clinical syndromes depending on predominant symptoms. These syndromes include behavioral variant (bvFTD), semantic dementia (SD), and progressive nonfluent aphasia (PNFA). 34 In bvFTD, patients show profound changes in personality and behavior, exhibiting apathy and disinhibition. Patients are often socially withdrawn and tend to engage in socially inappropriate behaviors such as shoplifting, making inappropriate or offensive remarks to others, and physical assault. 25 Repetitive motor behaviors, hoarding, overeating, and hyperorality are also common. 25 Patients lack insight concerning their disease. 34 Cognitive impairment tends to be less pronounced than the behavioral changes in bvFTD. 25
Patients with SD present with a fluent, anomic aphasia and often have asymmetric degeneration of the anterior temporal lobes. 34,35 Patients with left-sided atrophy lose semantic knowledge about words, objects, and concepts. 25 Right-sided atrophy yields a behavioral syndrome similar to bvFTD. 36,37 Compared with patients with bvFTD, patients with right-predominant SD are more rigid, have distinct compulsions, and are more likely to have weight loss, sleep disorders, and sexual dysfunction. 25 In both right- and left-sided SD, the disease spreads to the contralateral temporal pole, and patients develop symptoms corresponding to the resulting atrophy after a mean of 3 years. 37
The PNFA is a disorder of motor speech and language expression and is associated with left perisylvian atrophy. 34,38 Patients display motor speech deficits and often speak slowly or have trouble initiating speech. They also have difficulty producing and comprehending grammar. Reading is nonfluent and writing is agrammatic. 25
Currently, there are no disease-modifying treatments available for FTD. The only therapies that exist are for the control of symptoms and behaviors. Some limited data suggest that the selective serotonin reuptake inhibitors (SSRIs) can promote improvement in behavior and stereotypical movements. 39 –41 When patients have severe behavioral symptoms that are refractive to SSRI treatment and are dangerous (such as agitation or psychosis), the atypical antipsychotics olanzapine, 42 risperidone, 43 and aripiprazole 44 are sometimes indicated. Although these agents are riskier in elderly patients, the physician and family must weigh the risks and benefits of these agents in all patients with dementia. The cholinesterase inhibitors rivastigmine, 45 galantamine, 46 and donepezil 47 have been used with mixed results and do not appear beneficial in treating FTD. There is some suggestion that memantine may improve apathy, agitation, and anxiety. 48
A number of experimental therapies for FTD are in development. Tau-directed therapies include inhibition of tau kinases, inhibition of tau fibrillation, manipulation of tau-processing pathways, and inhibition of tau expression. 25 Because haploinsufficiency of PGRN results in FTD, another possible treatment strategy is increased expression of wild type PGRN. Also, pathologic TDP-43 is hyperphosphorylated, ubiquitinated, and cleaved to generate C-terminal fragments, and each of these steps represents a potential drug target. 25
Kufs’ Disease
The neuronal ceroid lipofuscinoses (NCLs) are a group of lysosomal storage diseases that cause neurodegeneration. The incidence of NCLs is 1 in 12 500 live births. 49 Ten subtypes, designated CLN1-10, have been described. While most of the subtypes occur in childhood, CLN4, also called Kufs’ disease, is an adult-onset form of NCL that causes early-onset dementia.
The genetic factors contributing to the development of Kufs’ disease are poorly understood. Of the 50 cases in a review, 16 appeared sporadic and 34 appeared familial; the majority had autosomal recessive inheritance. 50 It was recently discovered that the mutations in DNAJC5 cause adult-onset NCL. 51,52
In Kufs’ disease, as in all the NCLs, there is an accumulation of abnormal lipopigments, which form lysosomal inclusion bodies in neurons and other cells. 53 On histopathological analysis, storage bodies are found in characteristic fingerprint, curvilinear or granular osmiophilic patterns, and are required for diagnosis 54 (Figure 1). The clinical features of Kufs’ disease include dementia, seizures, gait abnormalities, and cerebellar and extrapyramidal symptoms. Two subtypes exist: type A and type B. Patients with type A Kufs’ disease present with new-onset generalized tonic–clonic seizures, usually at about 30 years of age. 50 Symptoms that follow include dementia, ataxia, progressive myoclonus epilepsy, and later, pyramidal and extrapyramidal signs. 55 Features of type B Kufs’ disease include dementia and behavioral abnormalities, sometimes associated with ataxia, motor dysfunction, and extrapyramidal and suprabulbar signs. 56
No effective treatments exist for Kufs’ disease. Antiepileptic agents may be useful for treating seizures. For the other NCLs, strategies such as enzyme replacement therapy, gene therapy, cell-mediated therapy, and pharmacological/small molecule intervention are being attempted. 49 Perhaps when a better understanding of the etiology of Kufs’ disease has been achieved, some of these strategies may be tested.
Niemann-Pick Disease Type C
Niemann-Pick disease is a group of autosomal recessive lysosomal lipid storage disorders. Four subgroups, types A through D, exist. Niemann-Pick disease type C (NP-C) is a cause of EOD. From 1988 to 2002, in France, United Kingdom, and Germany, the prevalence of NP-C is estimated to have been 0.66 to 0.83 per 100 000 individuals. 57
Niemann-Pick disease type C results from mutations in NPC1 and NPC2. A mutation in either gene will cause the disease. 58 –61 Genetic testing can detect mutations in both of these genes. In all, 95% of individuals with NP-C have mutations in NPC1 and 5% have mutations in NPC2. The NPC1 gene codes for a glycoprotein that associates predominantly with late endosomes. 62 The NPC2 gene codes for a small soluble lysosomal protein that binds cholesterol with high affinity. 59 In NP-C, cells display impairment in the processing and utilization of endocytosed cholesterol. As a result, GM2 and GM3 gangliosides accumulate in neurons. 59 Meganeurite formation, growth of ectopic dendrites, and neurofibrillary tangles may be found. Neuroinflammation and neuroaxonal dystrophy also occur. Ultimately, neurons die, especially the Purkinje cells of the cerebellum. 59 In addition, foam cells and sea blue histiocytes are usually found in the bone marrow. 59 Foam cells may be present in other tissues (Figure 1).
The clinical presentation of NP-C is highly variable. Age of onset varies from the perinatal period until the seventh decade of life. 59 Life span ranges from a few days to more than 60 years of age. 59 Systemic disease, when present, precedes neurological disease in all cases. Systemic manifestations include hepatosplenomegaly and, rarely, lung involvement. 59 Neurological features include progressive dementia, ataxia, dysphagia, dysarthria, and vertical supranuclear gaze palsy. Other common neurological manifestations include seizures, dystonia, cataplexy, and in late stages, psychiatric disturbances. 59
Current therapies for NP-C include antiepileptic agents for seizures, anticholinergic agents for dystonia and tremors, and physical therapy for spasticity and contractures. Miglustat has been shown to promote improvement in neurologic parameters and stabilization of neurologic disease. 63,64 A number of studies have been carried out in mice with imatinib, curcumin, non-steroidal anti-inflammatory drugs, steroids, and 2-HP-β-cyclodextrin. 59
Differential Diagnosis and Workup
As in all cases of cognitive decline, reversible causes must first be ruled out. Vitamin B12 and folate deficiencies, hypothyroidism, infection, electrolyte abnormalities, intracranial hemorrhage, intracranial mass, normal pressure hydrocephalus, and immune-mediated CNS diseases are treatable causes of cognitive decline. Appropriate laboratory and imaging tests should be performed to rule out these conditions. Medications should be reviewed for agents that may impact cognition. When medication is the culprit, cognitive symptoms often occur shortly after new medications are started or with an increase in dose. Also, depression and other psychiatric disorders may present with cognitive alterations. For example, patients with depression with psychotic features often have impaired cognition. A recent case report showed that cognition improved in such a patient when depression was treated with electroconvulsive therapy. 65 A history of prior depression or psychotic disorder warrants to the consideration of a treatable psychiatric diagnosis. Patients should be asked about psychiatric history and recent symptoms. If the workup for reversible causes of dementia is negative or there is a family history of EOD, patients should be evaluated by a neurologist and neuropsychological testing should be considered. A simplified diagnostic algorithm may be helpful (Figure 2).
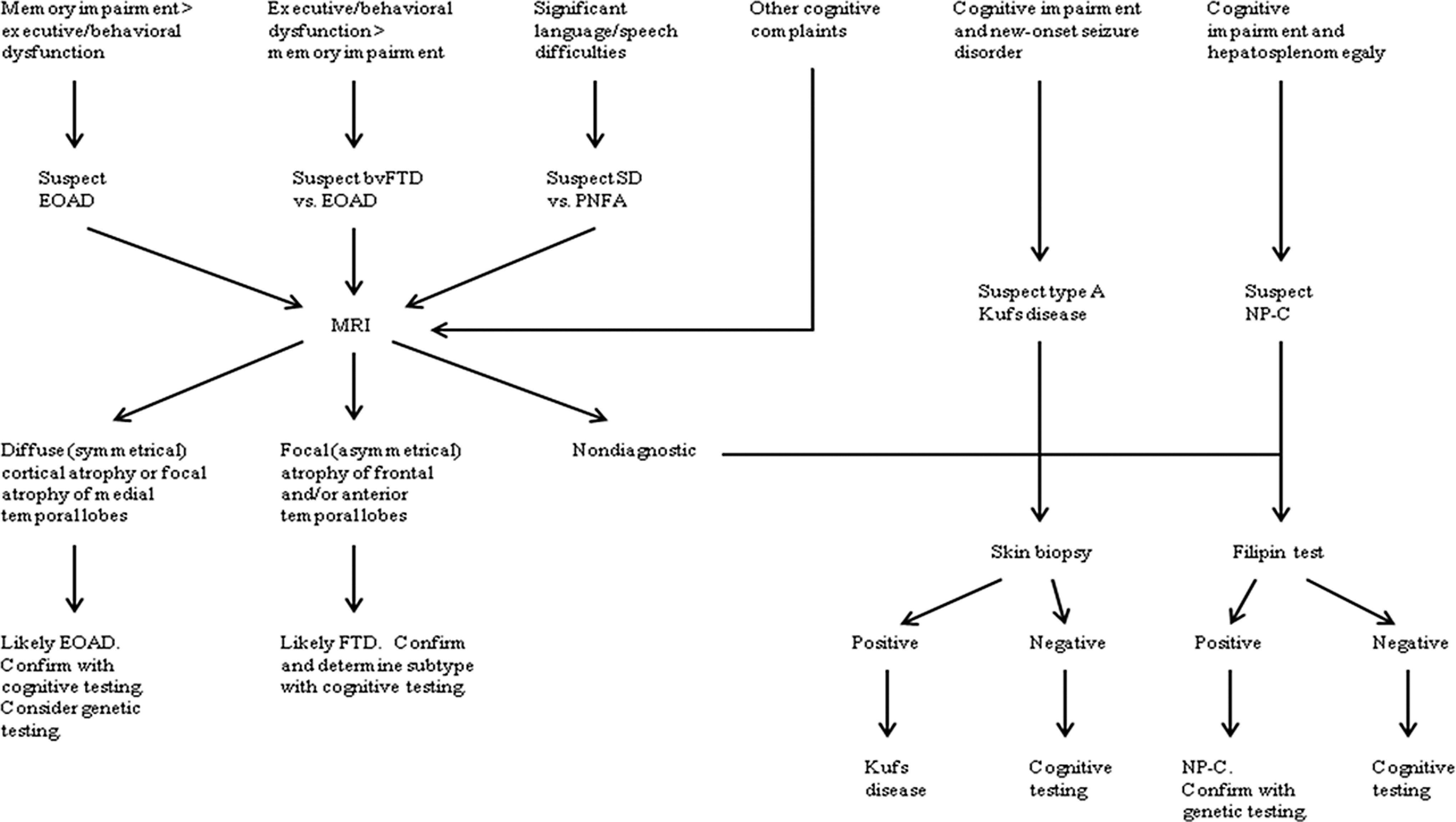
Algorithm for diagnosis of early-onset dementia. After reversible causes are ruled out, workup should be based upon the patient’s presenting symptoms.
The diagnosis of probable AD is a clinical diagnosis that requires a detailed history of the patient’s course of symptoms. Important findings include insidiously worsening episodic memory impairment and problems with social and occupational functioning. Because patients with AD usually lack insight concerning their disease, it is important to interview another source, such as a family member or caregiver. Cognitive testing reveals impaired memory formation, often with little benefit from cues. Other areas of cognition including executive functioning, visuospatial skills, language, and orientation are also involved, but to a lesser extent in AD. Neuropsychological testing is an integral part of the diagnostic workup. A finding of cortical atrophy on magnetic resonance imaging (MRI) is supportive. 66 Clinicians must remember that patients with EOAD often present initially with atypical manifestations such as behavioral and executive dysfunction, and patients presenting with memory impairment are often depressed or apathetic. Detailed family histories should be obtained to determine the mode of inheritance.
Usually, FTD can be distinguished from AD when behavioral or language abnormalities occur with intact memory. Executive function is often worse than memory loss—the opposite pattern of AD. Patients with FTD often exhibit frontal release signs and Parkinsonism, further aiding the diagnosis. However, EOAD may be confused for FTD when memory difficulties present in FTD or when frontal or behavioral features occur in AD. In such cases, neuropsychological testing and structural and functional brain imaging are useful. 30 In FTD, MRI of the brain often shows focal (asymmetrical) atrophy of the frontal and/or anterior temporal lobes. In AD, the tissue loss is more symmetrical and diffuse or localized to medial temporal lobe regions (Figure 3).
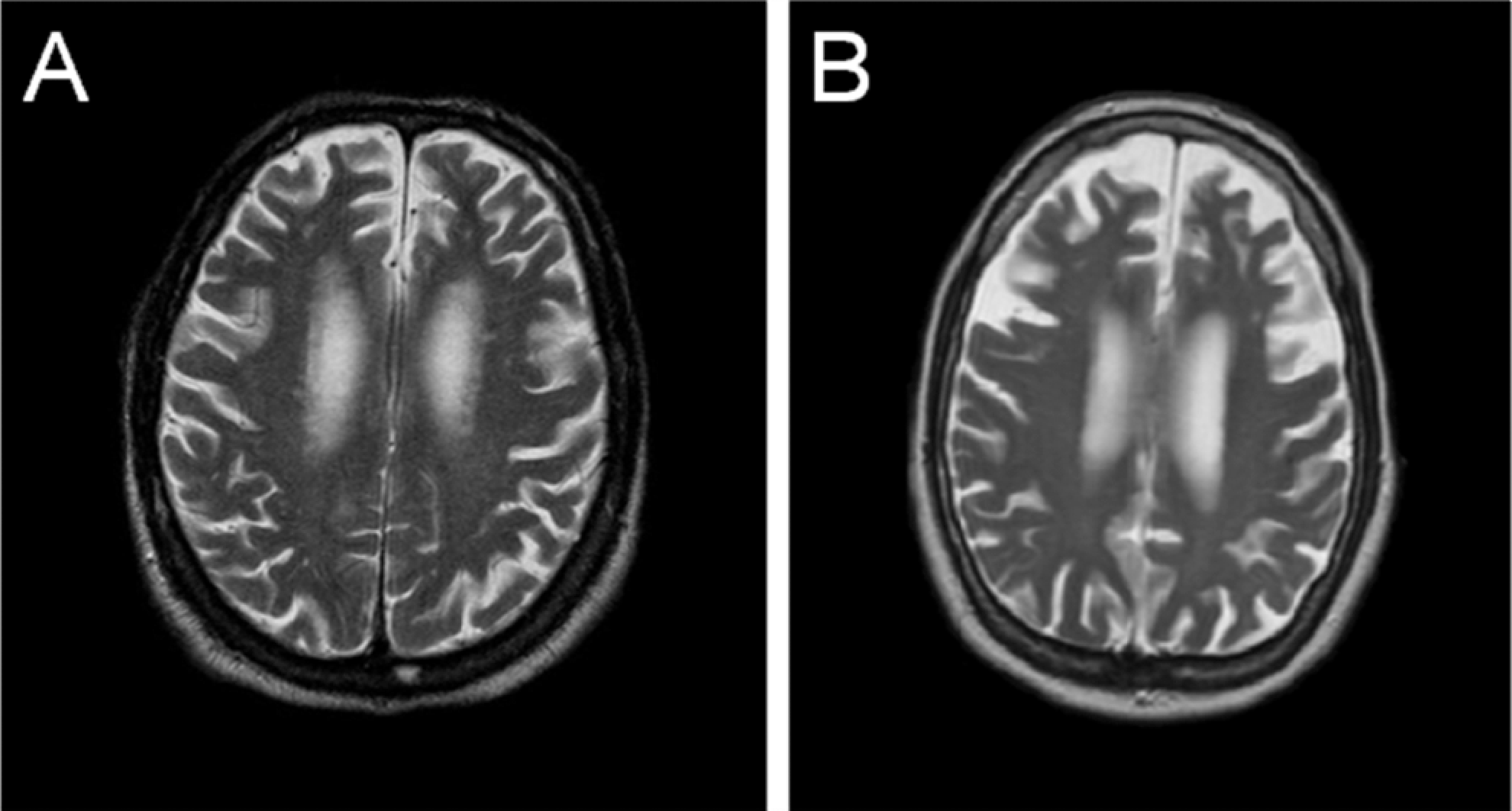
Comparison of MRI findings of AD and FTD. Images were obtained from MRI T2 sequences in the axial plane performed in a patient with AD (A) and FTD (B). Cortical atrophy is diffuse in the patient with AD, while in the patient with FTD, it is localized to the frontal and temporal lobes. MRI indicates magnetic resonance imaging; AD, Alzheimer’s disease; FTD, frontotemporal dementia.
Seizures are a prominent and early feature of type A Kufs’ disease, and patients who present with new onset of seizures and dementia should be evaluated for this condition. Because the primary manifestations of type B Kufs’ disease are dementia and behavioral abnormalities, it is possible that type B Kufs’ disease could be confused with bvFTD or EOAD. Brain imaging and neuropsychological testing can help clarify the diagnosis. If testing for Kufs’ disease is desired, a 3-mm punch biopsy deep enough to include sweat glands is recommended; biopsies revealing lipopigment accumulations are diagnostic.
Systemic manifestations of NP-C, such as hepatosplenomegaly, distinguish it from other forms of dementia. However, in some cases, only neurological symptoms are present. The ophthalmologic examination is especially helpful because often the earliest neurological sign is abnormal saccadic eye movements. 59 In patients who are suspected of having NP-C, a proposed algorithm for NP-C diagnosis recommends that a skin biopsy be performed and, if positive, sequencing of NPC1 and NPC2 genes be completed. 64
Conclusion
Dementia may result from a number of disease processes. The etiology can be difficult to determine because, particularly in younger patients, different causes of dementia may produce similar constellations of symptoms. Family histories may help the physician narrow the differential diagnosis. Some important causes of early-onset inheritable dementia include early-onset AD, FTD, Kufs’ disease, and Niemann-Pick disease type C. Table 1 summarizes the genetic causes, symptoms, and workup for each of these pathologies. Currently, treatments for these diseases are limited to the control of symptoms. However, disease-modifying therapies are in development and may one day slow, halt, or even help to reverse disease progression. As we await these advancements, it is important that we refine our diagnostic approach so that patients may be diagnosed correctly and treatment outcomes may be optimized.
Summary of Genetic Etiology, Symptoms, and Workup of Causes of Early-Onset Dementia
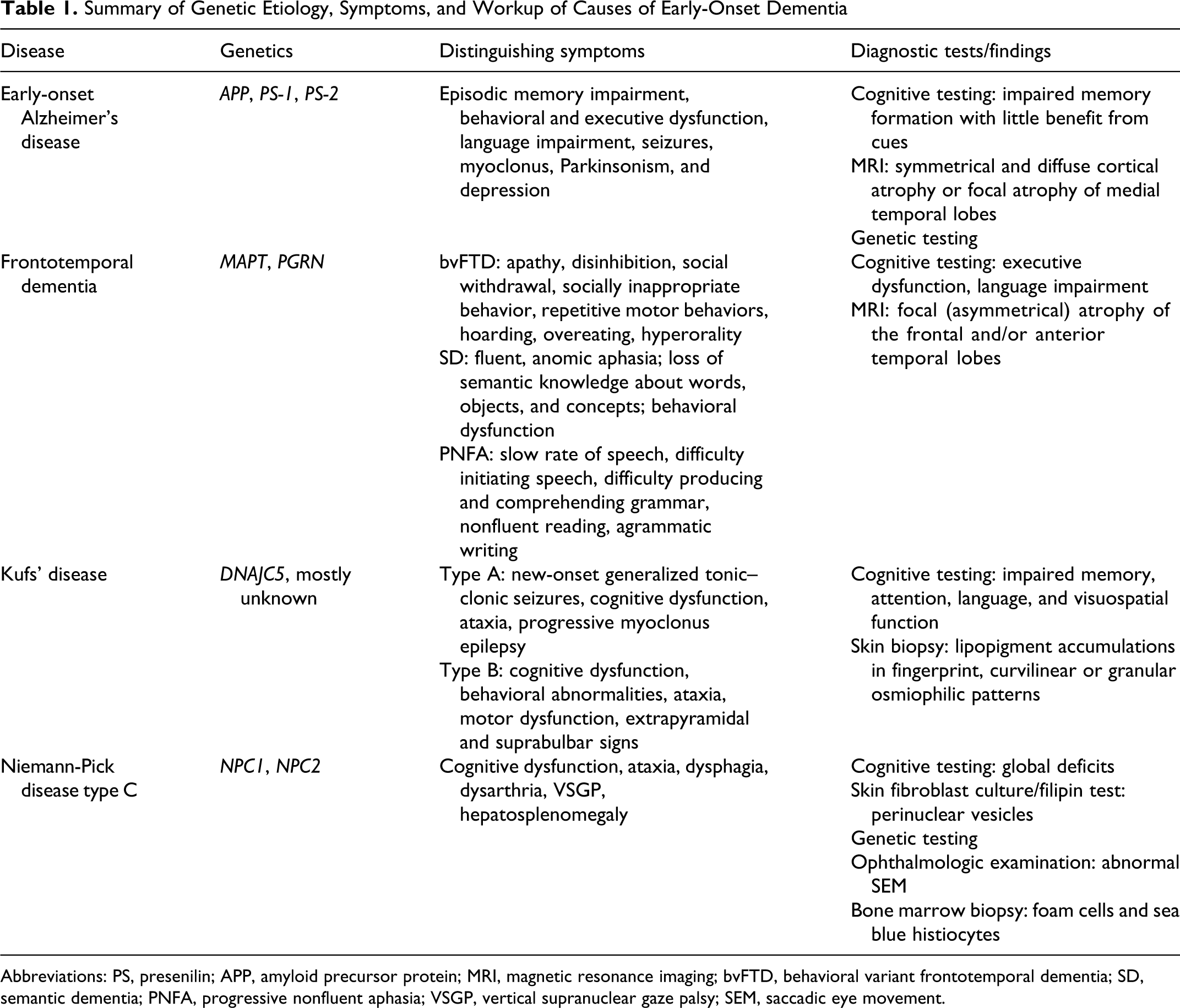
Abbreviations: PS, presenilin; APP, amyloid precursor protein; MRI, magnetic resonance imaging; bvFTD, behavioral variant frontotemporal dementia; SD, semantic dementia; PNFA, progressive nonfluent aphasia; VSGP, vertical supranuclear gaze palsy; SEM, saccadic eye movement.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.