
Abstract
This article reviews the significance of the microbiome in cancer epidemiology, mechanistic and technical challenges in the field, and characterization of the microbiome in different tumor types to identify biomarkers of risk, progression, and prognosis. Publications on the microbiome and cancer epidemiology were reviewed to analyze sample collection and processing, microbiome taxa characterization by 16S ribosomal RNA sequencing, and microbiome metabolite characterization (metabotyping) by nuclear magnetic resonance and mass spectrometry. The analysis identified methodology types, research design, sample types, and issues in integrating data from different platforms. Aerodigestive cancer epidemiology studies conducted by different groups demonstrated the significance of microbiome information in developing approaches to improve health. Challenges exist in sample preparation and processing (eg, standardization of methods for collection and analysis). These challenges relate to technology, data integration from “omics” studies, inherent bias in primer selection during 16S ribosomal RNA sequencing, the need for large consortia with well-characterized biospecimens, cause and effect issues, resilience of microbiota to exposure events (requires longitudinal studies), and expanding studies for fungal and viral diversity (most studies used bacterial 16S ribosomal RNA sequencing for microbiota characterization). Despite these challenges, microbiome and cancer epidemiology studies are significant and may facilitate cancer risk assessment, diagnosis, and prognosis. In the future, clinical trials likely will use microbiota modifications to improve the efficacy of existing treatments.
Introduction
Infectious agents including bacteria and viruses have been linked to cancer because they produce toxins, carcinogenic metabolites, and cause chronic inflammation. The number of malignancies associated with infectious agents represents approximately 20% of all cancers. Technological advancements have made it possible to examine the entire microbiome in different organs as a functional entity. Data generated during the past decade have demonstrated the discovery of novel microbiota that perform different functions in the body. At times, these microbiota are associated with the development of cancer. 1 –6 Variations in host responses to the microbiome are not studied extensively. Recent technological advancements also have made it possible to follow human exposure to environmental and dietary factors and study the role of microbiota in human health. The National Institutes of Health’s Human Microbiome Project (HMP) analyzed 4788 specimens from 242 healthy phenotyped adults selected from a cohort of 300 individuals. 7,8 From this group, 131 individuals were followed longitudinally to collect data from different anatomical sites to generate a reference microbiome. The HMP represents the normal microbiota of healthy adults (individuals free of any clinically diagnosed disease) in Western populations. Individuals were followed to analyze the microbiota at different habitats to understand the extent of individual variations and relationships among microbes. Such information could be useful in understanding microbe-associated diseases including cancer and in developing treatment strategies. Both the number and abundance of diverse microbes constitute an individual’s microbiome. To follow the microbiome of an individual, a researcher needs to follow the microbiome profile at different times. Clarifying the role of microbiota at an organ site necessitates moving beyond taxonomic overrepresentation and examining changes in the cancer-associated microbiome in a more functional context. Following up metabolomics flux may be useful in characterizing the functional microbiome. The association of cancer phenotype with specific microbiota has been largely based on observation studies with criteria such as the strength of association, including its consistency, specificity, and temporality. 9 Several studies have raised the issue of the cause and effect of the microbiome on health and disease. 10,11 Careful longitudinal studies should be conducted to distinguish between the association or cause (and consequences) of a disease due to the microbiota. The most common factors that affect the microbiota of an individual include genetic, epigenetic, dietary, stress-related, and depression-related factors.
16S ribosomal RNA (rRNA) sequencing and metagenomic profiling information should be valuable in developing personal and precision medicine approaches. Although interesting associations of microbiota with cancer development have been reported, the underlying etiology is not completely understood.
The most diverse microbes are found in the oral cavity and stool, and the least diverse microbes are present in the vagina. Microbiota diversity is measured in terms of alpha (within) and beta (in between) diversity. No taxa were universally present in the different habitats (anatomical sites) of an individual. Less dominant taxa are highly personalized in individuals and body habitats. Different habitats exhibit relationship-influencing physical features such as oxygen, moisture, pH, host immunological factors, and microbial interaction. In contrast to taxa, several metabolomic pathways showed limited diversity in different habitats. The functional identification of uncharacterized pathways will be essential in evaluating the role of microbiota in disease development.
Over a lifetime, each human develops a densely populated microbiome, and a change in population structure may result in altered microbiota. 12,13 The term “dysbiosis” is used frequently in microbiomics to indicate disruptions in the microbiota. The role of dysbiosis in inflammatory cancer and other diseases has been reported. 14 –16 Differences in microbiota composition exist in different organ sites, and colon microbiota has been studied the most in cancer development. 17,18 Colonic mucosa are in constant direct contact with microbiota and substances (eg, different metabolites and toxins, superoxide radicals) produced by them. Effects of diet on colon cancer development have been studied by several investigators. 19,20 Dietary components and their metabolites may contribute to polyp formation and further oncogenic processes. 21,22 Induction of proinflammatory responses by mucosa may contribute to carcinogenesis.
Although the field of the microbiome and cancer epidemiology has gained importance in recent years, mechanistic and technological challenges remain. Evaluating these challenges is the main focus of this article. The article discusses the microbiome in different aerodigestive tumor sites, unique features of the microbiome at these sites, and the current status of cancer epidemiology.
Research Design and Methodologies
Sample Collection and Processing
Sample collection and processing procedures vary and may affect microbiota analysis. Flores et al evaluated the effects of collection media and stool sample storage at 3 different temperatures and durations. 23 Microbiota analysis indicated stability of samples for beta-diversity but the loss of some rare taxa in samples that were not stored quickly at a low temperature. 23 In this study, samples were frozen in dry ice immediately (time = 0) or frozen at −80°C after 3 and 7 days. All samples were collected in triplicate. Ideally, samples should be kept frozen until further processing.
Cultivable and Uncultivable Microbes: Development of New Technologies and Culture Strategies
Some microbes, especially oral bacteria, are easy to collect but difficult to culture for further characterization. 24 New technologies and culture strategies are needed so that the catalog of cultivable oral bacteria can be expanded by capturing species that are currently uncultivable. Approaches such as different culture media, techniques requiring coculturing with various microbial partners, and the use of host-derived cells and factors could be worth testing. Studies should be conducted to answer questions such as what makes one species more easily cultivable than another or why one closely related species is cultivable while a genetically similar strain is uncultivable. These studies will help investigators understand whether domesticated isolates require compensatory mutations or other genomic rearrangements to adapt to in vitro culture conditions and if some reversible mechanism allowed a previously uncultivable isolate to revert back to an uncultivable state. Further development of molecular labeling, biofilm imaging, and metabolite detection is needed to discover and quantify metabolic interactions between community members and to demonstrate their ability to monitor these interactions in real time during long-term culture conditions.
Determining whether changes in microbiota are a cause of disease in studies that compare the composition of the microbiota in diseased individuals with that of healthy persons also is of interest. As a disease develops over several years, however, it becomes difficult to determine whether changes in the microbiota are a consequence of alterations (eg, in diet, physiology, environmental exposure) or whether they are causative.
Microbiome Taxa Characterization by 16S RNA Sequencing and Analysis
The most common method for characterizing the microbiome is to conduct a 16S RNA analysis 25 via 454 pyrosequencing. 26,27 A metagenomic survey is conducted using a hypervariable region of the highly conserved and universal 16S RNA gene as a phylogenetic marker. Bacterial taxa are identified based on sequencing results, and sequences are clustered into operational taxonomic units, followed by different assignments according to the Greengenes databank (http://greengenes.lbl.gov/cgi-bin/nph-index.cgi). The functional microbiome is characterized further by PICRUSt (http://picrust.github.io/picrust/), a bioinformatics software package used to predict the metagenome functional content of marker gene surveys and full genomes. Metagenomics information is useful in characterizing the composition of the microbial community. In characterizing gene-specific alterations in disease, samples that are used to delineate phylogeny using 16S rRNA can be used to amplify other genes from the microbial community by polymerase chain reaction (PCR), with degenerate primers targeting conserved regions of specific genes of interest. Expression of this PCR-amplified gene can provide insights into a particular function or type of metabolism in the microbial community. 28
Types of Methodologies Used
In the analysis presented in this article, most of the studies involving the microbiome and cancer epidemiology focused on specific cancers or specific infectious agents. Pancreatic, colorectal, and gastric cancers were the most studied cancers for which the microbiota were characterized. The most common methods for microbiome analysis were PCR 25 and sequencing, 27 but a few studies used metabolomics profiling approaches that identified cancer-associated short-chain fatty acids and specific sugars. 29 Furthermore, metabolomic profiling was useful in identifying potential pathways in cancer development. Both mass spectrometry (MS) and nuclear magnetic resonance technologies were used for metabolite characterization of the microbiome.
Research Design
Identification of specific microbes in the microbiodata as biomarkers requires an appropriate study design. Retrospective case–control studies can be conducted, but these studies cannot determine the temporality of disease-specific modifications with regard to disease occurrence. In cancer development, it is very difficult to distinguish “drivers” and “passengers,” and this prohibits the identification of early markers of the disease. Prospective studies can be useful in identifying disease biomarkers. Difficulties with prospective studies, however, include that they require large numbers of cases and controls and can be expensive to conduct. The results of prospective studies can be validated by small retrospective studies. The problem of field defect, which is associated with many such studies, can be resolved by taking healthy tissue samples close to the affected tissue.
Types of Samples Used and Commonly Investigated Viruses and Bacteria in Different Cancers
Saliva, stool, blood, and plasma were the common samples used in microbiome and epidemiologic studies. The following viruses and bacteria were commonly observed in different cancers: JC virus—breast, colorectal, and bladder cancers 30 –32 ; BKV polyomavirus—breast cancer 30 ; Chlamydia trachomatis—cervical cancer 33 ; human papilloma virus (HPV)—cervical cancer 34 ; Helicobacter pylori—gastric cancer 35 ; Epstein–Barr virus—lymphoma 36 ; cytomegalovirus—prostate cancer 37 ; Chlamydia pneumoniae—lung cancer 38 ; Merkel cell polyomavirus—skin cancer 39 ; and hepatitis B and C viruses—liver cancer. 40
Metatranscriptomics, Metaproteomics, and Metabotyping (Microbiome Metabolite Characterization)
Metatranscriptomics identifies genes that are expressed in a microbial community. 41 Metaproteomics characterizes microbial community functions that can be measured by MS to identify proteins that are present in a sample. 42 Metabolomics measures metabolic changes that occur in the microbiota. 43 In an organ site, released and consumed metabolites can be quantified to elucidate the interaction between microbiome and host metabolism. This may lead to the identification of diagnostic biomarkers. Fecal and blood metabolomics data were used to illustrate the predictive power of the Community and Systems-Level Interactive Optimization toolbox in analyzing metabolic interactions between the diet, gut microbiota, and host metabolism in a study by Shoaie et al. 44 This diet intervention study used data from obese and overweight individuals. Such models may facilitate understanding microbiome association and disease causality. Based on metabolite characterization and rRNA sequencing, the association between the oral microbiome and pancreatic cancer was demonstrated successfully by Michaud and Izard. 3
Data Integration
New approaches are needed to effectively mine the wealth of microbiota sequence data and integrate them with clinical and epidemiological metadata. Data sets, especially metagenomic data coupled with demographic and clinical indicators, are heterogeneous, sparse, and multidimensional. Applying unsupervised computational learning and interpretable association rule mining may resolve these problems. In studying colorectal cancer, 2 different epigenetic interaction networks using chemical–gene, disease–gene, and protein–protein interaction data from multiple sources were used. The results indicated a strong link between colorectal cancer and levels of trimethylamine N-oxide, which is a gut microbial metabolite of dietary meat and fat. 45 Mathematical modeling was used to develop such tools that can predict interaction of microbiome with diet and host metabolism. The model was validated, and findings from gut microbiota, diet, and metabolites were successfully integrated. 44 Such studies provide an opportunity to develop gut microbiome-dependent diagnostic tests and therapeutics for this deadly cancer.
Aerodigestive Cancers
The literature search on microbiome and cancer epidemiology is shown in Table 1. The data were generated using different terms, and the number represents the number of publications using these terms. Although the field of cancer microbiome and epidemiology is new, still a sizable number of studies conducting these studies were observed. Criteria for selecting a publication in cancer epidemiology field have been previously described in detail. 46 In this article, the author has discussed about microbiome associated with aerodigestive cancers. The main outcome of the analysis is presented below.
Literature Search on the Microbiome and Cancer Epidemiology.a
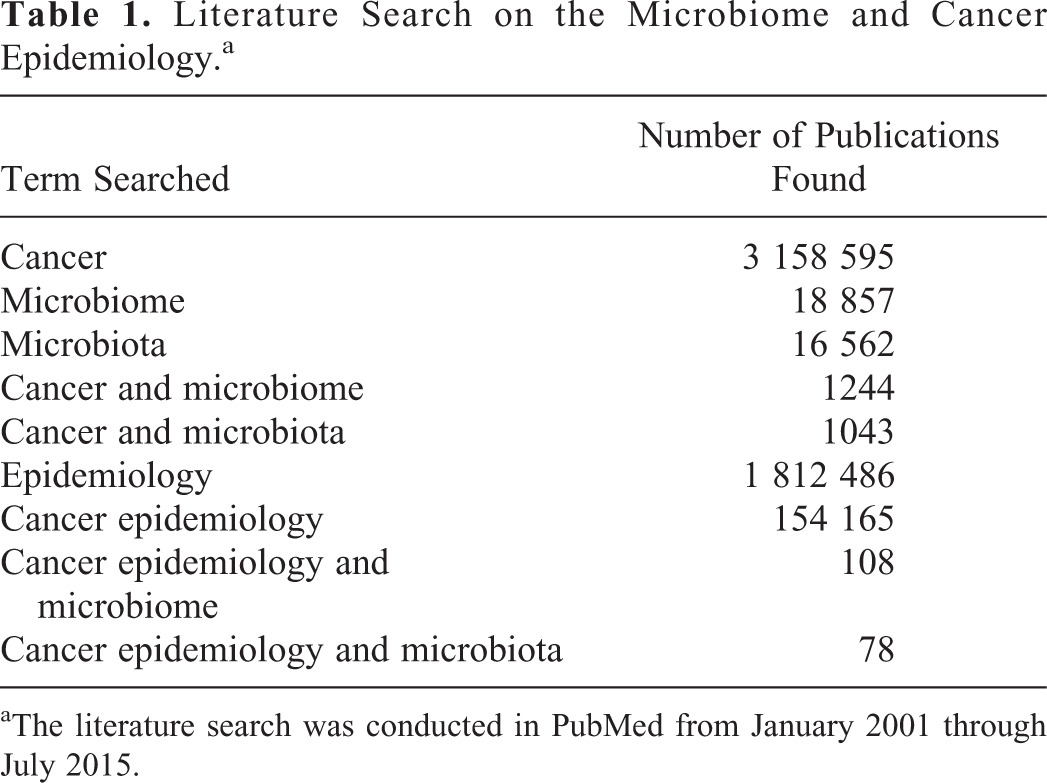
aThe literature search was conducted in PubMed from January 2001 through July 2015.
The role of bacteria in oral cancer has been proposed by different investigators, although the complete etiology of this cancer remains unknown. 2,47,48 The oral cavity harbors bacteria, fungi, viruses, protozoa, and archaea. The most common bacterial phyla present in the oral cavity are Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria. Most of the differences in the microbiomes of patients with oral cancer and healthy individuals occur at the species and strain levels. 49 In an epidemiologic study, oral cancer biospecimens contained more Exiguobacterium oxidotolerans, Prevotella melaninogenica, Staphylococcus aureus, and Veillonella parvula bacteria compared to their control counterparts. 50 In another study, analysis of the microbiome in swab samples from healthy individuals and patients showed almost double the number of anaerobes in patient samples. 51 Mager et al demonstrated the utility of the number of bacteria in saliva as an indicator of oral squamous cell carcinoma. 52
Cervical cancer, 53,54 oral cancer, 55,56 and head and neck cancers 57,58 have one common feature of the presence of HPV. 53,55 –60 Almost all patients with cervical cancer have HPV, whereas this virus is present in most of oral cancer cases and few head and neck cancer cases. More than 100 strains of HPV have been reported to date, although 2 strains, HPV16, and HPV18, are the most virulent among all strains. The number and titer of this virus influence microbiome of the organ site where this virus is present. From the health care point of view, the HPV vaccine has been successful in preventing cervical cancer, but for other cancers, results are awaited. 61 –63
Gastric cancer is the second leading cause of cancer-related mortality and fourth most common cancer across the globe. A complex community of noncultivable microorganisms has been reported in patients with gastric cancer, although the most prominent and well-studied bacteria in gastric cancer is H pylori. 1 H pylori infection induces a host immune response that causes gastric inflammation, epithelial atrophy, and dysplasia. 64 Only a few studies have investigated the community of bacteria and gastric cancer. One epidemiologic study showed that regular consumption of fried foods, spicy foods, large amounts of salt, and large amounts of fat were associated with gastric cancer in a population located in Hehuang Valley, China. 65 In another study, H pylori infection was associated with an increased risk of gastric cancer, mainly noncardia gastric cancer in Japan. 66 This group suggested that smoking cessation and dietary modification may prevent this form of H pylori-associated gastric cancer. However, H pylori infection has been associated with a lower risk of esophageal cancer, probably due to reduced acid reflux and epithelial damage in those tissues. 67,68 Esophageal cancer is a serious malignancy with regard to mortality (sixth among all cancers worldwide) and prognosis. Its incidence is expected to increase during the next 10 years. Human papilloma virus infection also has been reported in esophageal cancer. 69 Investigators also have proposed the involvement of a complex microbiome in cancer in the distal esophageal region. 70 The microbiota of the upper digestive tract were found to be associated with cancer-predisposing states in esophageal and gastric cancers. 71
Pancreatic cancer is one of the most lethal cancers; more than 90% of patients with pancreatic cancer die within 5 years. The pancreatic cancer microbiome was analyzed to understand the etiology of this cancer and identify markers for its early detection. Risk factors for pancreatic cancer include smoking, chronic pancreatitis, obesity, and type 2 diabetes. Because all of these factors affect the host immune system, it was logical to study the role of the microbiome—especially bacteria—in the carcinogenesis of pancreatic cancer. Three epidemiologic studies were conducted to evaluate the relationship between periodontal diseases and pancreatic cancer. 72 –74 A positive association was observed in all 3 studies, and in one of these studies, a 4-fold increase in the risk of pancreatic cancer was observed among patients with severe periodontitis. Elevated levels of antibodies against Porphyromonas gingivalis were also observed. 74 Farrell et al compared the microbiota content in the saliva of healthy patients and cases with pancreatic cancer and observed that 2 bacteria, Neisseria elongate and Streptococcus mitis, could distinguish between cases and controls with 96% sensitivity and 82% specificity. 75 The metabolomics of pancreatic juice were similar to those of the oral microbiota, and a few oral microbe-specific metabolites were detected in the pancreatic juice.
Worldwide, colorectal cancer is the third most commonly diagnosed cancer and the fourth leading cause of cancer death. Gut microbiota, with 10 to 100 trillion microorganisms, alter inflammatory tone, insulin signaling, lipid accumulation, and production of short-chain fatty acids that are involved in food intake. Different species of Fusobacterium were reported in colorectal cancer, especially F nucleatum, F mortiferum, and F necrophorum. 76 Another group reported finding Clostridium leptum, C coccoides, and Faecalibacterium prausnitzii in colorectal cancer tumor samples. 77 Enrichment of select bacteria, such as F nucleatum, has been implicated in the development of colorectal adenomas and adenocarcinomas. 5,78,79 Dietary fiber can favor the growth of butyrate-producing bacteria, which have a beneficial effect on gut microbiota. 80 One epidemiologic study compared the composition of gut microbiota in European children and rural African children; a significant enrichment of Bacteroidetes and depletion of Firmicutes were observed. 81 African children had high fiber content in their diet compared to European children, and the difference in microbiota composition between the 2 populations could be due to their different diets. In a model system, it was demonstrated that switching from a low-fat, plant-rich diet to a Western diet altered the microbial composition and metabolomic pathways. 82 A bacterial driver–passenger model for colorectal cancer has also been proposed. 83 In one study, gut microbial activity was measured by determining metabolite profiling. 84 The gut microbiota are considered an important effector in the relationship between diet and cancer and have a potential role in cancer prevention.
Exposure of the lung to bacteria, such as Mycobacterium tuberculosis and pneumonia-causing bacteria, is known. Not many epidemiologic studies, however, have been conducted that demonstrate an association between the microbiota and the lung cancer. The lung microbiota react with and metabolize environmental xenobiotics. In one case–control epidemiologic study, lung cancer microbiota were studied in women in China who were exposed to cooking smoke. 85 Buccal cells and saliva were used for microbiome characterization. The results indicated that the composition of the microbiota in exposed women was different than in unexposed women. 85 Lung cancer cases also had a decreased abundance of Spirochaetes and Bacteroidetes phyla compared to controls, and oral samples from cases had more Firmicutes than controls. M tuberculosis is considered a risk factor for lung cancer in both smokers and nonsmokers. 86
Discussion
Both the number and abundance of diverse microbes constitute an individual’s microbiome. To follow the microbiome of an individual, a researcher needs to follow the microbiome profile at different times. Clarifying the role of microbiota at an organ site necessitates moving beyond taxonomic overrepresentation and examining changes in the cancer-associated microbiome in a more functional context. Following up metabolomics flux may be useful in characterizing the functional microbiome. The association of cancer phenotype with specific microbiota has been largely based on observation studies with criteria such as the strength of association, including its consistency, specificity, and temporality. 9 Several studies have raised the issue of the cause and effect of the microbiome on health and disease. 10,11 Careful longitudinal studies should be conducted to distinguish between the association or cause (and consequences) of a disease due to the microbiota.
Below are key conclusions that cover the current status of cancer microbiome and epidemiology.
Conclusion 1: There Are Problems With Sample Preparation and Storage Methods, and There Is a Need to Incorporate Microbiomes of Viruses, Fungi, and Role of Exposure Events
Characterization of early microbiome assembly and maturation is needed to understand the role of the source inoculum, host immune system, diet, and environment. There is a need to develop tools and algorithms that can integrate 16S sequence information and metagenomics with metatranscriptomics and metaepigenomics.
Analyses are needed for the functional properties of microbiomes, along with more reference strains and functional analyses of the strains. The roles of viruses, phages, and microbial eukaryotes in the microbiome are still not clear. Strain-level diversity and strain-specific metabolites and products should be determined and characterized.
Redundancy (overlap of function among multiple species) and resilience (resistance to, and capacity to recover from, perturbation) are observed frequently in different microbiomes. Attempts have been made to investigate the basic mechanisms that mediate the resilience of prominent gut microbiota during inflammation. 87,88 One of the mechanisms includes lipopolysaccharide modifications and increased AMP resistance triggered by pathogen-induced inflammation. 87,88
Robust bioinformatics environments and computational tools are needed—especially infrastructure and tools that can operate with terabases and pentabases of microbiome data and that also can analyze different data types (generated in multiomics studies). The fundamental differences between sequence analysis of the genome of a single microorganism versus metagenome are that the sequence reads from the DNA of a single microbe can be assembled, aligned, and annotated easily (where a reference database exists), whereas most microbial genomes are closed, circular structures that is difficult to annotate and characterize. In colon cancer, distorted microbial activities affect gut epithelium leading to inflammation and colorectal cancer. Random forest tree searching with rule learning may be applied to analyze and interpret big data generated in such studies. In colon cancer, distorted microbial activities affect the gut epithelium, leading to inflammation and colorectal cancer.
Better methodologies and approaches are needed. For example, a microbiome signal may be difficult to separate from a human signal, and stool is not representative of the gut tract. Additional PhyloChip microarrays should be developed for further characterization of microbiome.
Conclusion 2: Two Major Unknowns Are Temporality of Findings and Sources of Bias
Bias in determining the microbiome composition has been reported, mainly due to different DNA extraction protocols and DNA amplification, although other factors such as sequencing artifacts, DNA copy number, sampling depth, and primer design also contribute to bias during 16S sequencing. 89 This bias can be reduced by analyzing mock communities comprised a prescribed proportion of cells from several relevant bacterial strains. Mock communities—artificial microbial communities created by mixing known quantities of bacterial isolates, DNA clones, or PCR products—generally are used for quantifying bias. 90
Conclusion 3: Utility of Cohorts Is Essential for Microbiome and Cancer Epidemiology Studies
Although many microbiomes and their distribution in different populations have been demonstrated, the fundamental mechanics that guides the assembly of specific microbes is not completely understood. There is a need for multiple prospective cohorts with phenotypes for host and microbiome associations. Cancer epidemiology cohorts (CECs) are large, observational human population studies with thousands of study participants in which groups of people with a set of characteristics or exposures are followed systematically and prospectively for the incidence of new cancers. Those CECs that collect information about microbiome also can be used to evaluate whether the effect of exposure contributing to cancer development is reflected in the composition of microbiota. 91 Such studies can be further expanded by conducting transcriptomics and metabolomics for functional characterization of altered microbiome. The cohort-based study design is a mainstay of epidemiologic inquiry because of its many advantages over other epidemiologic study designs. These advantages include unbiased assessment of multiple exposures prior to the onset of cancer (such as serologic biomarkers or risk behaviors) and the ability to assess multiple outcomes. Recently, different composition and functional differences in microbiome were reported in a number of cohorts. 92 During the past 2 decades, CEC-based studies have facilitated the unraveling of the complex etiology of cancer and provided fundamental insights into key environmental, lifestyle, and genetic determinants of this complex disease. Findings from these cohort studies are critical for risk prediction analyses and models. Such results also may serve as a basis for cancer control measures and prevention practices for at-risk groups and populations and have been useful in providing the basis for the design and testing of many preventive and therapeutic interventions. Cohort biorepositories already support genomic and epigenetic studies and in the future could support proteomic and metabolomic studies. Such studies should be done to characterize the microbiome.
Looking Ahead
New strategies such as fecal transplantation and antibiotic, prebiotic, and probiotic approaches have shown promising results in treating disease. 93 Transferring the intestinal microbiota is a possible treatment, and this approach is a part of personalized medicine. Treatment is continued in patients who show a positive response to such alterations in their microbiota (reestablishment of the healthy microbiota). Brandt et al described a multicenter clinical follow-up study in which the fecal transplantation approach was successfully applied in treating bowel disease. 94 Other aspects that have not yet been investigated are the relationship of the early microbiome to the microbiome in health and disease throughout life and the relationship between diet and the microbiome in populations of non-European ancestry and in non-Western diets, customs, and practices. The microbiome and cancer epidemiology field should now move to human health and disease. Future research projects should include diverse populations in order to circumscribe and associate the functional properties of the microbiome with other features of these populations. Research resources such as the cohort consortia should be utilized more to plan large studies to understand cancer etiology. Association studies should be evaluated for causalities. Based on the discussion above, it is fair to say that the microbiome has tremendous potential in understanding cancer etiology and for developing interventions and therapeutic targets.
Footnotes
Abbreviations
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.