
Abstract
Hepatocellular carcinoma (HCC) is the primary form of liver cancer. It causes ∼ 800 000 deaths per year, which is expected to increase due to increasing rates of obesity and metabolic dysfunction associated steatotic liver disease (MASLD). Current therapies include immune checkpoint inhibitors, tyrosine kinase inhibitors, and monoclonal antibodies, but these therapies are not satisfactorily effective and often come with multiple side effects and recurrences. Metabolic reprogramming plays a significant role in HCC progression and is often conserved between tumor types. Thus, targeting rewired metabolic pathways could provide an attractive option for targeting tumor cells alone or in conjunction with existing treatments. Therefore, there is an urgent need to identify novel targets involved in cancer-mediated metabolic reprogramming in HCC. In this review, we provide an overview of molecular rewiring and metabolic reprogramming of glucose metabolism in HCC to understand better the concepts that might widen the therapeutic window against this deadly cancer.
Introduction
Hepatocellular carcinoma (HCC) accounts for over 90% of primary liver cancers and is the fourth leading cause of worldwide cancer-related death. 1 HCC is a rapidly increasing cancer frequency known to be a health disparity in the United States by incidence, stage, and survival. 2 The burden of HCC varies remarkably with sex, and the incidence rates are higher in males than females and individuals over 55 years of age. 2 Likewise, the incidence and mortality from HCC varies by race and ethnicity in the United States, with non-Hispanic Asians and Pacific Islanders topping the list. 2 The majority of the HCC subtypes develop in the context of chronic liver damage and inflammation where viral hepatitis caused by the Hepatitis B virus (HBV)/Hepatitis C virus (HCV) is the primary cause. 3 However, recently, the incidence of HCC as a direct result of nonviral origins has been rapidly increasing, especially in developed countries. 4 In the United States, which has a high rate of obesity and metabolic syndrome, many of these individuals also suffer from metabolic dysfunction associated steatohepatitis (MASH), a severe form of metabolic dysfunction associated steatotic liver disease (MASLD). 5 MASLD is characterized by liver steatosis, inflammation, hepatocellular injury, and fibrosis, 5 and it has become the primary causative liver disorder with a high predisposition to HCC. 6 There is an increasing trend, with < 20% of HCC being attributed to MASLD in the United States, which makes it a tremendous scientific concern that requires intensive research. 7
Metabolic reprogramming is a process by which cancer cells alter their metabolism to support rapid growth and proliferation. One hallmark of HCC, like any other cancer, is the metabolic adaptation to enhance glucose metabolism to produce energy and the building blocks needed for cell growth and proliferation. 8 It is also thought to provide a survival advantage to HCC by allowing the cancer cells to proliferate despite low oxygen levels and nutrients. 9 Importantly, this rewired glucose metabolism (glucometabolism) may provide potential targets for treating and managing HCC. This review covers aberrant glucose metabolism, called glucometabolic adaptations in HCC, and a brief account of its microenvironment, which may be exploited for therapeutic intervention. The schematic representation of these significant metabolic changes and reprogramming is depicted in Figure 1 and discussed in the following subsections of this review.
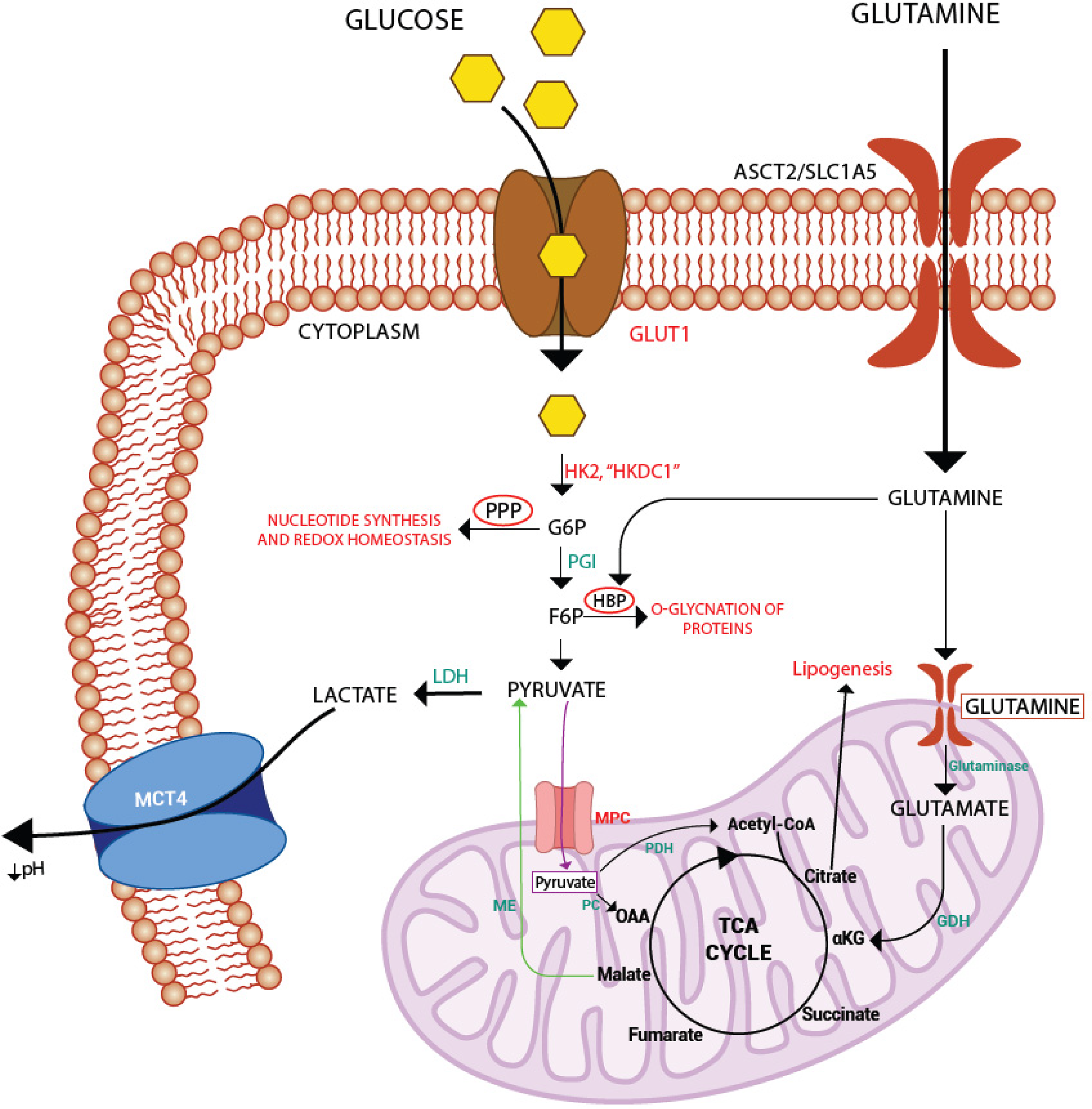
Reprogramming of glucose metabolism in HCC: the HCC cells adopt several altered metabolic processes to adapt, survive, and proliferate in the tumor-hostile environment. Here are some main such pathways schematically represented. Red color denotes tumor promotive metabolic reprogramming, green denotes some HCC-specific pathways/metabolites, and thick black arrows represent the upregulation of pathways in HCC.
Glucose Uptake
Glucose transport is one of the significant events responsible for glucose uptake and trapping of glucose inside cells, and it depends on glucose transporters (GLUTS).10,11 GLUTs are a family of facilitative glucose transporters involved in regulating glucose uptake and metabolism, and nearly all family members have been confirmed in the liver. 12 However, the significant GLUTs in normal hepatocytes are thought to include GLUT1, GLUT2, GLUT5, GLUT8, and GLUT9. 12 In HCC, GLUT1 mRNA is overexpressed by 68.2%, which correlates significantly with invasiveness and metastasis. 13 The enhanced expression of GLUT1 in HCC and its lower expression in corresponding normal and benign hepatic tissue suggests that this transporter is involved in the increased glucose uptake by HCC cells, acting as a tumor-promoting target with prognostic and diagnostic significance. 13 GLUT2, the major isoform in normal hepatocytes, is crucial in glucose sensing, and hepatic glucose output is also upregulated in HCC and is positively correlated with poor prognosis in patients. 14
Glycolysis
Enhanced glycolysis in HCC provides energy in the form of ATP for tumor progression and, more importantly, feeds biosynthetic pathways such as pentose phosphate pathway (PPP), hexosamine biosynthetic pathway (HBP), and tricarboxylic acid (TCA) cycle, essential for cancer progression. 15 After cancer cells have achieved enhanced glucose uptake, the next significant step is the need to trap it inside due to the bidirectional transporting capabilities of the GLUTS, and this is where the role of hexokinases (HKs) comes into play. HKs are the first rate-limiting enzymes involved in glycolysis, which phosphorylate glucose, thus trapping it inside the cells. 16 Five HK isozymes (HK1-4 and HKDC1) have been identified in humans with diverse tissue distribution and enzyme kinetics. HK4, commonly known as glucokinase, is the predominant HK isozyme of hepatocytes. 16 However, a switch from GCK to HK2 has been observed in HCC, which correlates with cancer progression. 17 This effect is observed despite oxygen availability and is not a consequence of the physiological response to hypoxia as expected at the tumor core owing to poor vascularization. 18 Another switch recently observed in HCC is GCK to HKDC1, which interacts with mitochondria, leading to HCC progression. 19 HKDC1 is a novel fifth hexokinase previously implicated in gestational diabetes, regulation of whole-body glucose metabolism, and insulin sensitivity. 20 The overexpression of HKDC1 has also been reported in gastric cancer, 21 lung cancer, 22 endometrial cancer, 23 breast cancer, 24 nasal cancer, 25 and colon cancer. 26 The upregulated HKDC1 in HCC correlated with proliferation and poor prognosis of patients. 19
Several other glycolysis-related enzymes are upregulated and implicated in HCC progression, notably PFK-1, GAPDH, and PK.27,28 Phosphofructokinase-1 (PFK-1), which catalyzes the conversion of fructose 6-phosphate (F-6-P) to fructose 1,6-bisphosphate (F-1,6-BP) using ATP, is another rate-limiting and highly regulated enzyme of the glycolytic pathway. 29 Three isoforms of this enzyme have been identified in mammals: PFK-M (found in muscle), PFK-P (found in plasma), and PFK-L (found in the liver). In humans and rats, expression levels of PFK-L, PFK-M, and PFK-P subunits are expressed in this descending order. 29 A fully functional PFK1 is a tetramer whose formation and stabilization largely influence the glycolytic flux rate. 29 It has been shown that increased PFK-1 activity promotes glycolysis and proliferation in cancer cells. 30 PFK1 is under the allosteric control of fructose-2,6-bisphosphate (F-2,6-BP), a product formed from fructose 6-phosphate through catalysis by PFKFB3 (6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3), which increases PFK1 activity.31,32 Thus, PFKFB3 mediates pro-glycolytic shift in HCC proliferation by the allosteric activation of PFK1, and its overexpression confers tumor aggression in HCC.33,34
Surprisingly, GAPDH (glyceraldehyde-3-phosphate dehydrogenase), often used as an internal control in immunoblotting and quantitative PCR experiments, is frequently upregulated in HCC, enhancing glycolysis and tumor progression. 35 GAPDH also affects glycolysis indirectly by regulating the mammalian target of rapamycin complex 1 (mTORC1) signaling pathway, which promotes the “Warburg Effect.”36,37 The mTORC1 pathway is a growth signaling mechanism that is hyperactivated in HCC, where it promotes rapid cancer cell growth, survival, and invasion, 37 and GAPDH regulates mTORC1 signaling according to glucose availability (ATP). 37
Pyruvate kinase (PK) is the third and final rate-limiting and irreversible enzyme that catalyzes the last step of glycolysis and is also found to have increased expression in HCC.38,39 Four isoforms of this enzyme have been identified in mammals and designated as PKM1, PKM2, PKL, and PKR based on their tissue distribution. 39 PKM1 isoform is found in tissues such as muscle, heart, brain, and others with a high catabolic need. PKL is found chiefly in the liver and PKR in RBCs. PKM2 is the embryonic form associated with several cancers.39,40 It has also been shown that switching from PKL (liver-specific isoform) to PKM2 promotes enhanced glucose uptake, glycolysis, and increased oxidative stress in hepatocarcinogenesis. 41 Furthermore, PK also regulates the expression of HIF-1α and Bcl-XL expression in HCC. 41 Conclusively, the deranged glycolysis is perhaps the major metabolic rewiring in HCC, providing cancer cells a suitable niche for enhanced growth and progression.
Pentose-Phosphate Pathway
This shunt pathway of glycolysis (Figure 1) generates: (a) ribose-5-phosphate needed for nucleotide synthesis, which is then used to synthesize genetic material and high-energy molecules such as ATP required for cell division, and (b) reducing equivalents such as NADPH required for lipogenesis and maintaining the redox balance of the cell.42–44 It has been shown that most enzymes of the PPP are upregulated in HCC. 45 In particular, the expression of glucose-6-phosphate dehydrogenase (G6PDH), the rate-limiting enzyme of the pathway, is highly upregulated in HCC and is associated with migration, invasion, poor prognosis, and chemoresistance. 46 G6PDH also undergoes O-GlcNAcylation mediated by HBP (another rewired metabolic process in HCC, detailed in the following subsection), enhancing its activity and stability and thus augmenting its tumorigenic potency in HCC. 47 The PPP has 2 functionally interrelated arms, the oxidative and the nonoxidative, with separate roles. The oxidative arm mainly produces the reducing equivalent, NADPH, while the nonoxidative branch produces pentose phosphates and other metabolites, such as fructose-6-phosphate (F6P) and glyceraldehyde-3-phosphate (G3P).48,49 The cell conditions that remarkably affect this shunt are high division rates and the need for reducing equivalents. 49 An important observation in this regard is that hepatocyte proliferation, ie, regeneration of the liver after specific stimuli (physical, chemical, nutritional, or virus-induced liver injury), does not seem to involve the oxidative branch of PPP. 50 Consequently, there is no significant change in the expression of G6PDH in the normal proliferating hepatocyte. 46 Thus, it appears that the primary role of the oxidative arm of PPP is to maintain redox homeostasis in HCC, which is burdened by high intracellular reactive oxygen species (ROS) relative to healthy tissue. 48 This rewired metabolism may also provide an effective therapeutic interventional target for treating HCC by specifically downregulating the oxidative arm of the PPP. However, any advancement in this regard for HCC is yet to be realized.
Hexosamine Biosynthetic Pathway
This metabolic shunt pathway diverts 2% to 3% of total glucose carbons from glycolysis and has received attention lately for its role in glucometabolic reprogramming. 51 HBP diverges at the third step of glycolysis (Figure 1), where glutamine-fructose-6-phosphate aminotransferase 1 (GFAT1), the rate-limiting enzyme of HBP, converts F6P and glutamine to glucosamine-6-phosphate and glutamate. 51 The final product of HBP, uridine diphosphate N-acetylglucosamine (UDPGlcNAc), is the donor for O-linked β-N-acetylglucosamine modification (O-GlcNAcylation) of many proteins on their Ser/Thr residues commonly referred to as O-linked glycosylation (O-GlcNAcylation), thereby modulating their functions by enhancing stability and reducing protein turnover. 52 The enzyme O-GlcNAc transferase (OGT) catalyzes this transfer, and aberrant O-GlcNAcylation has been implicated in HCC. 53 The expression of GFAT1 was upregulated in HCC cell lines, leading to increased tumorigenic proliferation and migration in vitro. 54 Expectedly, high expression of GFAT1 correlates with poor prognosis in HCC patients. 54 In another study by Cao et al, 55 OGT expression was significantly elevated in HCC cells relative to normal hepatocytes. In another systematic study, global O-GlcNAcylation remarkably increased HCC tumors compared to healthy tissues. 56
Increased O-GlcNAcylation can also lead to tumorigenesis and metastasis by modifying oncogenic transcription factors such as YAP and CHK2 (checkpoint kinase 2). 53 Receptor for activated C kinase 1 (RACK1) is stabilized upon O-GlcNAcylation mediated by HBP, which then interacts with and activates protein kinase C βII isoform, leading to downward phosphorylation and activation of targets such as eukaryotic translation initiation factor 4E (eIF4E) to translate oncogenes. 57 Moreover, this initiation factor can also undergo O-GlcNAcylation, enhancing its stability and protection from proteasomal degradation, thereby promoting HCC progression. 55 Another mechanism of O-GlcNAcylation-mediated HCC progression is the inactivation of the tumor suppressor Rab3A, which is inactivated upon O-linked β-N-acetylglucosamine modification. 58
Another fate of the UDP-GlcNAc is in N-linked glycosylation, where the final product of HBP forms one of the essential carbohydrate building blocks of the conserved Glc3Man9GlcNAc2 oligosaccharide transferred during the ER phase of glycosylation. 59 The N-linked glycans ultimately affect the protein activity and function, resulting in changes in cellular signal transduction, adhesion, and migration.60,61 In HCC, the N-glycosylation patterns correlate with genetic subtypes. 62 The HCC biomarker alpha-fetoprotein (AFP) is now being screened for aberrant N-glycosylation in addition to expression levels for early and effective diagnosis. 63 N-glycosylation of the cluster of differentiation 147 (CD147) at Asn 152 also affects its folding and stability, promoting tumor metastasis in hepatocellular carcinoma. 64 Overall, HBP-mediated glycosylation of proteome is correlated to HCC progression and could open novel avenues for therapeutic intervention for HCC by targeting this pathway.
Fate of Pyruvate and Lactate
The regulation of pyruvate, the end-product of glycolysis in the mitochondria, is another critical factor determining anabolic versus catabolic glucometabolism. Pyruvate is catalyzed to acetyl CoA by pyruvate dehydrogenase (PDH) in normal cells, thus linking glycolysis to the TCA cycle or fermented to lactate/lactic acid (LA) in the cytoplasm under hypoxia by lactate dehydrogenase (LDH). 65 However, HCC cells reprogram themselves, where pyruvate dehydrogenase kinase 1 (PDK1) is upregulated and activated, which inactivates PDH, suppressing the TCA cycle and shifting glucose metabolism towards glycolysis.66,67 Another important factor involved in the fate of pyruvate in HCC is the mitochondrial pyruvate carrier (MPC) responsible for the pyruvate transport into the mitochondria for the oxidative phase of cellular respiration. 68 Downregulation of the MPC complex in HCC has been observed, thus inhibiting pyruvate transport to mitochondria and shifting glucometabolism to enhanced lactate production. 69
Additionally, high levels of LDH-A mRNA have been found in HCC, suggesting elevated lactate levels, which can also be ascribed to increased LDH-A enzyme levels. 70 This switch reduces the oxygen dependency of HCC cells and promotes glycolysis persistently.71,72 However, accumulation of lactic acid could lead to an acidic environment and subsequent apoptosis of HCC cells. So, on the one hand, rapid glycolysis may provide sufficient energy to the proliferating HCC cells, but on the other hand, the accumulation of LA would mean their death. 73 To circumvent this, HCC cells upregulate monocarboxylate transporters (MCTs) to export accumulated lactate into the extracellular spaces. 74 MCT4 has been implicated in the HCC progression and spread, while MCT1 activates the Wnt/β-catenin signaling pathway to promote glycolysis in HCC cells.74,75 Thus, in HCC, lactate's fate is rewired to prevent accumulation, which keeps aerobic glycolysis running and prevents the tumor cells from acid buildup and apoptosis.
Gluconeogenesis
Gluconeogenesis is a “reversal” of glycolysis where glucose is synthesized from noncarbohydrate carbon sources such as glycerol, lactate, pyruvate, and glucogenic amino acids. 76 Metabolic reprogramming in HCC leads to inhibition of gluconeogenesis by downregulating the pathway’s key enzymes, thereby providing cancer cell plasticity and tumor growth advantage. 77 This is reflected in the altered expression pattern of some key enzymes of gluconeogenesis, such as phosphoenolpyruvate carboxykinase 1 (PCK1, also known as PEPCK-C), fructose-1,6-bisphosphatase, and glucose-6-phosphatase.77,78 PCK has 2 isoforms, PEPCK-C and PEPCK-M, distributed in the cytosol and mitochondria, respectively, and involved in decarboxylation and then phosphorylation of oxaloacetate to form phosphoenolpyruvate (PEP). 79 PCK1 has been implicated in its prominent role in cataplerosis. It acts as a tumor suppressor in the case of HCC, leading to imbalanced energy homeostasis by disturbing glycolysis and the TCA cycle, resulting in enhanced oxidative stress and energy stress. 79 However, HCC cells undergo molecular rewiring to downregulate PCK1/2 for their survival and progression.79,80 Fructose-1,6-bisphosphatase (FBP1) is another crucial and rate-limiting enzyme of gluconeogenesis and is found to have significantly reduced expressions in HCC. 81 This rewired expression of FBP1 has been listed with decreased gluconeogenesis and enhanced tumor progression in HCC. Moreover, HCC specimens exhibiting low expression of FBP1 had a highly malignant phenotype, including large tumor size, poor differentiation, impaired gluconeogenesis, and enhanced aerobic glycolysis. 81 Glucose-6-phosphatase (G6Pase) is another critical enzyme of hepatocyte carbohydrate metabolism and has high activity in a normal, healthy liver. However, HCC tissues were found to have significantly lower G6Pase activity, possibly because of the tumor's enhanced glucose intake and utilization.82,83 To sum up, gluconeogenesis is downregulated in HCC, and activating this pathway could be a potential treatment strategy for patients with HCC.
Anaplerosis and Cataplerosis
HCC cells adjust TCA cycle activity, providing metabolic plasticity to proliferate despite extraordinary genetic and environmental disadvantages. This is achieved by cataplerosis, where TCA cycle intermediates are transported away from the mitochondria to participate in biosynthesis, and anaplerosis, which replenishes TCA intermediates to maintain the stability of the cycle. 84 Anaplerotic enzyme pyruvate carboxylase supports the TCA cycle by supplying it with oxaloacetate and catalyzing pyruvate's carboxylation into oxaloacetate. 85 Knockdown of this enzyme resulted in the decreased flux of glucose-derived intermediates, reducing HCC proliferation. 85 The abnormal reliance on the anaplerosis of glutamine (Figure 1) is a characteristic of many malignancies that have long been known. 86 Here, we have discussed glutamine and acetyl-CoA as 2 essential representatives of anaplerotic and cataplerotic intermediates in HCC, providing the tumor with much-needed survival plasticity.
Glutamine Anaplerosis
In HCC, converting glutamine into α-ketoglutarate (α-KG) to maintain glucometabolic intermediates in a glucose-deficient tumor microenvironment (TME), is yet another reprogrammed metabolism adopted, especially during the metastatic stage. 87 This conversion involves deaminases, glutaminases, and glutamate dehydrogenase 1 (GLUD1).87,88 α-KG then drives the TCA cycle through anaplerosis in the nutrient-challenged HCC cells, providing metabolic plasticity and survival advantage to the HCC. 88 Also, K-glutaminase (GLS1) is upregulated in HCC during the epithelial–mesenchymal transition, converting glutamine to glutamate, thereby increasing the TCA intermediates without increasing the TCA enzymes. 89 This suggests the glutamine anaplerotic role in the proliferation, invasion, and migration of HCC cells. 90 It has also been observed that there is increased expression of glutamine receptor ASCT2 in HCC cells, leading to the increased uptake of glutamine from the plasma of HCC patients, thereby lowering their plasma glutamine concentrations. 91 Another protein of keynote importance concerning glutamine metabolism in HCC is glutamine synthetase (GS), which catalyzes the conversion of cellular glutamate to glutamine and is upregulated in HCC. 92 Apart from its role in anaplerosis described earlier, the high glutamine activates the mTORC1, which is associated with the further growth and proliferation of the liver cancer tissue. 93 Enhanced glutamine intake in HCC is also involved in the several biosynthetic pathways that enable these cancer cells to proliferate. 94 It acts as a nitrogen donor for synthesizing nucleotides and other amino acids such as arginine, aspartate, asparagine, serine, cysteine, glycine, and alanine. 94
Acetyl CoA Cataplerosis
As discussed above, the rapidly dividing HCC cells push for de novo lipogenesis to meet increasing energy demands, and acetyl-CoA acts as the precursor for the same. 95 Apart from β-oxidation of fatty acids and glutamine catabolism in cells, acetyl-CoA is also produced by mitochondrial oxidative decarboxylation of pyruvate derived from glycolysis.96,97 Therefore, metabolic rewiring of enhanced glycolysis in HCC ensures a continuous supply of this precursor for lipogenesis through cataplerosis. 98 Acetyl-CoA is also a substrate for histone acetylation, which is involved in the epigenetic regulation and reprogramming of genetic material in HCC. 98 Normal cells have a mechanism to hydrolyze abnormal acetyl-CoA and check cell growth through the enzyme acyl-CoA thioesterase 12 (ACOT12). 99 However, HCC rewires its metabolism by downregulating the ACOT12, resulting in metastasis and poor prognosis. 98 Downregulation of ACOT12 increases acetyl-CoA levels, leading to HCC metastasis by the epigenetic induction of genes involved in epithelial-to-mesenchymal transition (EMT). 98 Increased levels of acetyl-CoA have also been observed in other cancer types, including breast cancer, glioblastoma, and prostate cancer, indicative of the pro-metastatic role of this metabolite.100–102 Thus, anaplerosis and cataplerosis provide survival flexibility to HCC in the hostile environment of a tumor.
TME Alteration in HCC Contributes to Deranged Metabolism
The TME consisting of fibroblasts/stromal cells, lymphocytes, immune cells, vasculature, and extracellular matrix (ECM) plays a crucial role in cancer cell development, differentiation, survival, and proliferation.103,104 TME continuously evolves during cancer growth, providing a niche to support tumor growth and differentiation by helping cancer cells adapt to newer conditions. 105 TME also has growth factors and cytokines secreted by constituent cells and their organelles, forming a proper niche, which offers advantages to cancer cells in seemingly hostile conditions such as hypoxia, nutrient deficiency, and necrosis. 105 Due to increased glucose consumption, HCC cells induce glucose deprivation and hypoxia in the TME, leading to the expression of hypoxia-inducible factor 1 alpha (HIF-1α) as a response. 106 HIF-1α, in turn, mediates glycolytic metabolic alterations that drive cancer progression, resistance to therapy, 107 and poor prognosis to HCC. 108
There are a variety of stromal cells and inflammatory immune cells in TME, including cancer-associated fibroblasts (CAFs), macrophages, myeloid-derived suppressor cells (MDSCs), dendritic cells and regulatory T cells (Treg) exhibiting different functions compared to the non-cancer peripheral tissues. 109 HCC has been linked with tumor-associated stromal cells, especially hepatic stellate cells (HSCs), the primary subtype of stromal cells involved in liver fibrosis. 110 Most HCCs develop from severe liver fibrosis or cirrhosis, and the fibrotic liver facilitates the formation of a pro-TME. Since the pathway to HCC most often stems from fibrosis, it has been found that the otherwise quiescent phenotype, HSCs, are constitutively activated, which transdifferentiate into myofibroblasts with enhanced ECM production. 111 Another component of HCC TME is extracellular vesicles (EVs) secreted by cancer cells and stromal cells. They are essential for HCC malignancy and serve as biomarkers for early diagnosis.112,113 PKM2 containing EVs secreted by HCC cells has been found to reprogram glucose metabolism in monocytes, leading to their tumor-associated monocyte differentiation and eventually promoting HCC metastasis and progression. 114 It has also been observed that HCC cells release EVs containing various oncogenic micro-RNAs (OncomiRs), which are taken up by surrounding HSCs in the TME, leading to their differentiation into cancer-associated HSCs, thus jointly establishing the pro-tumor environment. 115 EVs derived from stromal cells set up a virtual environment for the growth, invasion, metastasis, and even drug resistance of HCC. 116 A recent study shows that EVs released from HSCs in the TME contain HK1, which the HCC cell hijacks, leading to its metastatic progression, 117 reflecting the rewired enhanced glucose metabolism in the HCC. Thus, EVs derived from cancer and stromal cells are considered some of the most important mediators of cellular crosstalk between tumors and the TME (Figure 2). HSC-derived EV containing HK1 is speculated to play a significant role in the metabolic plasticity of HCC cells. Overall, TME in HCC, like other solid tumors, promotes metastasis and malignancy.

TME and its interaction with HCC: crosstalk between stromal cells and HCC cells jointly establish the pro-tumor environment. After receiving appropriate signals from adjacent developing HCC cells, the hepatic stromal cells are transformed into cancer-associated cells in the TME, augmenting the HCC cell progression by supplying various pro-tumor materials such as enzymes and other factors, as detailed in the manuscript text.
Latest Trends and Challenges in HCC
The incidence rates of HCC have increased by 75% in the last 3 decades. 118 MASLD, formerly known as NAFLD, is an alarming public health issue worldwide with a predisposition to HCC. This term refers to a spectrum of liver maladies such as steatosis to metabolic steatohepatitis (MASH) (previously nonalcoholic steatohepatitis/NASH) to MAFLD-related cirrhosis and HCC. 119 Emerging scientific research is revealing some previously uncovered pathways in liver cancer research. For example, HKDC1 is the novel hexokinase recently identified to have a role in liver cancer progression. 19 This kinase is upregulated in MASLD and HCC. 19 In this study conducted in our lab by Khan et al, 19 it has been revealed that mitochondrial-bound HKDC1 regulates metabolism, proliferation, and survival of liver cancer (LC), and HKDC1-KO significantly affects glucose flux, energy metabolism, and mitochondrial function leading to less ATP production, thereby impacting cell-cycle progression and ER stress induction. The study provides another therapeutic opportunity for intervention by targeting this HK like other reprogrammed glucometabolic pathways. Since HKDC1 has nominal expression in normal hepatocytes but is highly upregulated in LC cells, novel small molecule and peptide-based inhibitors could target HKDC1, specifically its mitochondrial interaction in LC. The study also points out that targeting the HK1/HK2 as part of traditional HCC treatment is insufficient and probably the reason for the low prognosis in HCC/LC. In another study, 634 genes were consistently altered in HCC and are potentially relevant targets for onward studies in preclinical and clinical contexts. 45 Of these, there are 284 upregulated genes involved mainly in glucose metabolism, thus offering valuable therapeutic targets for HCC. However, HCC has biocomplexity and heterogeneity, which results in therapeutic challenges and added advantages for its survival. The scarcity of knowledge about the cellular origins of HCC, metabolic plasticity drivers, and therapeutic resistance is still a future challenge in HCC. These challenges must be considered in future directions in HCC research and treatment.
In conclusion, HCC is a complex cancer with diverse reprogramming, especially in glucose metabolism. The overview of the research findings should help understand the disease better and help find appropriate targets to cure cancers, especially LC/HCC, as it is still rising. In recent years, there has been an upward trend in the incidence rates of various cancers, especially HCC. This increase has spurred greater attention and resources dedicated to cancer research and the development of therapeutic interventions. However, it is important to note that interventions currently in place against HCC are not without significant limitations. As a highly documented fact, glucometabolic alterations in HCC account for its aggressive nature, progression, and metastasis. This review highlights the possible and appropriate targets offered by metabolically reprogrammed HCC cells, especially the enzymes of glucose metabolism, which can be combined with the existing therapies to have a lasting cure for this increasingly deadly disease. Thus, clinically meaningful and long-awaited improvements in overall survival for HCC patients with advanced and metastatic disease can be anticipated.
Footnotes
Abbreviations
Author Contributions
Conceptualization, MWK and SB; writing—original draft preparation, SB, ZF, HI, IC, and SB; writing—review and editing, MWK. All authors have read and agreed to the published version of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics Statement
This is a review article therefore, an ethics statement is not applicable.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the DOD Peer Reviewed Cancer Research Program (grant number W81XWH2010650).