
Abstract
Cancer is a public health problem that threatens human health. Due to the lack of specific and rapid diagnosis and treatment methods, the 5-year survival rate of patients has not been effectively improved in the past 10 years. Abnormal gene expression is closely related to the occurrence and development of cancer. Cancer diagnosis and treatment methods based on genetic testing have received extensive attention in recent years. It is essential to explore specific and rapid cancer genetic testing methods. Taking ovarian cancer as an example, we reviewed the progress of specific and rapid nucleic acid detection methods related to cancer risk assessment, low-abundance mutation detection, and methylation detection, to provide new strategies and ideas for related research.
Introduction
Cancer has become a public health problem that seriously affects human health and quality of life. In the United States alone, approximately 1 918 030 new cancer cases and 609 360 deaths are expected in 2022, according to the latest statistics. Ovarian cancer is a common malignant tumor in women. It is estimated that in 2022, there will be 19 880 new cases of ovarian cancer and 12 810 deaths in the United States. 1 The early symptoms of the cancer are hard to observe, and the existing inspection methods cannot detect cancer in an early stage. 2 About 70% of ovarian cancers are already in advanced stages when they are discovered. 2 There is an urgent need to develop new detection methods with good specificity and sensitivity for the early diagnosis and treatment of cancer.
The occurrence of cancer is closely related to abnormal gene expression. Gene mutations and promoter methylation in cancer driver genes can cause abnormal expression of the corresponding RNA and protein, leading to abnormal cellular functions. The relationship between abnormal gene expression and cancer has gradually become clear. For example, genes such as BRCA1/2, PTEN, MLH1, and MSH2 play essential roles in the occurrence and development of ovarian cancer. The detection of related mutated genes provides a significant reference for ovarian cancer genetic risk assessment, early screening, and prognosis assessment. In addition to genetic mutations encoding structural proteins, non-coding RNAs (ncRNAs) 3 and epigenetic changes 4 also provide us with a wealth of genetic information in the era of precision medicine. Therefore, genetic testing provides essential guidance for ovarian cancer screening, staging, and prognosis evaluation. For genetic testing, sequencing, polymerase chain reaction (PCR), and fluorescent probes are the most commonly used methods. Pyrosequencing, Sanger sequencing, next-generation sequencing (NGS), and other sequencing methods have become practical tools. 5 Besides, amplification methods, like selective PCR, and droplet PCR, further improved the sensitivity of the sequencing.6,7 Except for the methods of sequencing and PCR, nucleic acid probes have become a research hotspot in recent years due to their low cost and high specificity, providing a new strategy for mutation detection. 8 In the era of precision medicine, genetic diagnosis has become an essential method for early cancer diagnosis. We reviewed the progress from the following aspects: homologous recombination repair (HRR) associated gene mutation detection, cancer-associated low-abundance free DNA and circular tumor cell detection, ovarian cancer-related extracellular vesicle detection, ncRNA detection, and methylation detection methods. We hope this review can provide valuable targets for future cancer genetic detection and new ideas for detection method innovation.
HRR Associated Gene Mutation Detection
Gene mutation is one of the critical factors in the occurrence of ovarian cancer. HRR is complex progress for double-strand DNA breaks (DSB) and is highly related to BRCA1 and BRCA2. 9 On the one hand, BRCA mutations strongly correlate with ovarian cancer, and they contribute approximately 25% to ovarian cancer. 10 BRCA1 and BRCA2 mutations lead to the cumulative risk of ovarian cancer in women at age 80 of 44% and 17%, respectively. 11 High-risk populations with a personal or family history of ovarian cancer, breast cancer, fallopian tube, or peritoneal cancer should get tested for related mutations. On the other hand, BRCA mutations are strongly associated with homologous recombination deficiency (HRD). 12 HRD means the disfunction of HRR for DSB. The cancer cells with BRCA mutations cannot repair DSB, which provides the opportunity for applying the PARP inhibitor (PARPi) to improve the prognosis. Recent research suggests that HRR-related mutations are not limited to BRCA. Detection of CDK12, Fanconi Anemia genes, and other HR DNA-damage gene mutations is also essential for the application of PRAPi. Therefore, it is significant to detect HRR-associated gene mutation. Clinical detection of HRR-related mutations is mainly limited to the DNA level. As a methodological review, we take BRCA-related mutation detection as an example to briefly summarize the detection methods. Single-base mutations, small fragment insertions and deletions, and large genomic rearrangement (LGR) are the 3 most common forms of BRCA gene mutations.11,13 Commonly used methods for detecting gene mutations are sequencing, probes, and PCR.
The Methods of Sequencing and Probes
Representatives of commonly used sequencing methods are Sanger sequencing and NGS. For BRCA mutation detection, Sanger sequencing has become the gold standard for detection. However, there is a long working time (3-5 days) for the Sanger sequencing to detect the target. Besides, this method has missed the detection of LGR, and it needs to be combined with other approaches to complete the detection of LGR. 14 Compared with Sanger sequencing, NGS has higher detection throughput, lower detection limit, and shorter working time, which makes the revolution in biology and medicine. However, it is a more expensive and complicated procedure. The exaction of the nucleic acid, library preparation, and bioinformatics analysis are the main parts of the workflow. After obtaining a large amount of sequencing information, this method can obtain information about long fragments or even the entire genome through data analysis. NGS only can target part of the genome when detecting mutations. To enrich the target fragments, it has to combine PCR, molecular inversion probes (MIP), hybrid capture, and other methods. 15 Weren et al 16 combined a single-molecule molecular inversion probe (smMIP), single-molecule tagging, and multiple capture methods to analyze the open reading frames of BRCA1 and target sequence. It will extend to a complete chain under the action of the enzyme. Then PCR is used for amplification of the target. The probe (Figure 1A and B) comprises a recognition sequence and a stem loop. The enriched products can be directly sequenced when the probe binds to the target sequence with molecular markers. The smMIP method takes advantage of the feature that single-molecule tags can enrich low-abundance target segments, improving NGS analysis's sensitivity. Therefore, this method can analyze and detect low-quality DNA, such as the DNA from formalin‐fixed, paraffin‐embedded (FFPE) tissue. Although smMIP has better recognition specificity and lower cost, the length of its recognition region cannot be too long, or it will affect the binding efficiency of the probe and the target, which has a few limitations on its application. Buzolin et al 17 used the Ion Torrent PGM (Personal Genome Machine) platform to test the detection efficiency of NGS for BRCA1/2, and the results showed that the sensitivity of the NGS workflow test could reach 95.6% compared with the existing methods. Since the reliability of NGS detection of LGR depends on the library preparation method and bioinformatics analysis tools, NGS alone to detect LGR may have false-negative results. Because the target fragment needs to be amplified during the library preparation process, the PCR method has the problem of amplification bias. Moreover, the hybrid capture method has the problem of limited binding efficiency with the target fragment, so false-negative results are often produced, which affects the further use of NGS. To solve the problem, Daniela et al 18 developed a new method, CRISPR-DS (Figure 2), based on CRISPR-Cas9 and duplex sequencing (DS). This method uses the cleavage activity of the CRISPR-Cas9 system to generate fragments of uniform length, which are enriched by size selection. They demonstrated that the LOD could reach 0.1%. Multiplex ligation-dependent probe amplification (MLPA) is one of the usual methods of the probe. It is a standard and straightforward method for detecting LGR (Figure 1C). MLPA consists of 2 parts of probes with PCR primers. After specifically identifying and binding to the target sequence, the probe can connect to form a complete chain specifically and then be amplified and analyzed by PCR. This method is a high-throughput technology based on PCR, which can analyze up to 50 sequences in a single reaction. It can determine the relative copy number of the target sequence and detect the deletion/duplication of the target gene exon. Furthermore, the copy number of the target can be shown by the relative intensity of the amplified product. Sequencing methods are often combined with MLPA methods to detect LGR. Ruiz et al 19 used massively parallel pyrosequencing combined with MLPA to detect BRCA1/2 mutations in high-risk populations, and the approach has a specificity of 99.99%. However, due to the partial binding of the probe to exons, the ability of MLPA to detect LGR is limited. Combining the MLPA and NGS can improve detection efficiency and reduce the probability of errors. Besides, the combination of NGS and MLPA can reduce false-negative results in LGR testing. Kwong et al 20 combined NGS and MLPA to detect small nucleotide variations and LGR. Among 120 detected mutations, 8 mutation sites (Table 1) are checked out by MLPA but not by NGS.

Brief introduction of the scheme of smMIP and MLPA. (A) The basic structure of smMIP; (B) The workflow of smMIP; (C) The scheme of MLPA.

The basic workflow for CRISPR-DS.18 (A) Sequence selection by the size after the processing of CRISPR-Cas9. After selection, the target will connect with the double-stranded DS adapters. (B) Error correction by DS. Creat the single-strand consensus sequence (SSCS) reads by PCR. The SSCS reads from the same DNA molecule can compare with the other to become a double-strand consensus sequence (DCS). Only mutations detected in both strands can be counted as true mutations.
Eight BRCA Mutation Sites Detected by Kwong et al Using MLPA but not Detected by NGS.20

The Methods of PCR
The sequencing method can detect mutations more specifically and sensitively for BRCA mutation detection in the 2 application scenarios of genetic counseling and targeted drug evaluation. However, the shortcomings are also obvious: time consuming and expensive. Unlike sequencing, PCR is fast and cheap. The PCR method can detect single-base mutations, fragment deletions, and insertions at known sites. This method can also solve the problem that the sequencing method cannot effectively detect LGR. 21 PCR methods mainly include real-time quantitative PCR (rt-PCR), multiplex PCR, and high-resolution melting curve-PCR (HRM-PCR).
rt-PCR is a standard method for detecting tumor mutations. Based on this method, allele-specific real-time PCR (AS-PCR) commonly applies in clinical mutation detection research (Figure 3A). This method uses a primer 3′ terminal base to detect the mutation. Once the template strand matches the 3' end of the primer, then amplification can proceed; if the template strand has a mutation and mismatch with the 3' end of the primer, then amplification cannot continue. The AS-PCR process is simple and can detect low-quality DNA from pathological specimens. The detection limit of its common work platform ranges from 1% to 7%,22,23 but this method can only detect mutations at known sites. Multiple primers are used in the multiplex PCR method to amplify multiple fragments simultaneously, improving detection efficiency. Cini et al 24 analyzed 8 common high-frequency mutations of BRCA1/2 in the population by a series of PCR methods. They added an extensible probe designed according to the mutation site based on the multiplex PCR. The extension probe is only one base away from the mutation site, and they have tails of different lengths at different mutation sites. The tail lengths of probes for detecting different mutation sites are different. After adding ddNTP, a single base extension begins, then the extension process is terminated, and finally, the results are analyzed by electrophoresis (Figure 3C). HRM-PCR is a method that uses differences in length between sequences, GC ratios within the target, and base complementation to screen mutations after PCR. The detection time is short, and the cost is low. Besides, the entire reaction runs in a closed tube, and the possibility of sample contamination is less. 25 This method can detect deletions, duplications, and point mutations. To analyze mutations in all coding regions and intron-exon boundaries of BRCA1/2, Minucci et al 26 constructed a low-cost and high-throughput analysis method based on the HRM-PCR. The fluorescence peak height ratio of the BRCA1 fragment and albumin (ALB) fragment after HRM-PCR was used to evaluate the copy number change of BRCA1. Compared with the MLPA method, this method has 100% consistency. The HRM-PCR has high specificity, but the detection limit is low. Ihle et al 23 compared the detection limit of HRM-PCR with other methods for the same mutation sample and found that the detection limit of the HRM-PCR is 6.6% (Table 2), the same as the Sanger sequencing method but lower than NGS (2%). The advantages of PCR are simple and economical. It can complete mutation detection in application scenarios such as genetic risk consultation and targeted drug evaluation.

PCR method for evaluating genetic risk of ovarian cancer and related ctDNA detection. (A) The basic workflow of AS-PCR; (B) The workflow of BM-PCR; (C) the method of multi-PCR for 8 BRCA mutation sites detection.
Evaluation of Common Mutation Detection Methods by Ihle et al.23

Use cobas® as the working platform of AS-PCR.
Gene Detection Method for Early Diagnosis of Ovarian Cancer and Early Recurrence Intervention
In recent years, the discovery of a series of low-abundance mutations has provided new targets for the early intervention of cancer. They have become disease markers for early diagnosis and early detection of recurrence. Traditionally, a tissue biopsy is the mainstream method of cancer genetic testing, which means that relevant tissues are obtained through invasive approaches for testing to assist in diagnosis. In recent years, various genetic biomarkers of ovarian cancer have been discovered, such as cell-free DNA (cfDNA), circulating tumor DNA (ctDNA), circulating microRNA, and ncRNA (including but not limited to microRNA, lncRNA, circRNA). Genetic testing provides a powerful reference for early diagnosis of ovarian cancer, early detection of disease recurrence, prognostic evaluation, disease staging, and recurrence detection.
Detection of cfDNA and ctDNA
Cells release DNA into the circulatory system during their life activities. These circulating free DNA are called cfDNA. The cfDNA containing the mutant DNA fragments with tumor characteristics is circulating-tumor DNA (ctDNA), and the detection of ctDNA has become a feasible method for early diagnosis of ovarian cancer and early detection of ovarian cancer recurrence. The existing approaches for detecting ctDNA mainly include sequencing, PCR, and probe methods. As for the first-generation sequencing techniques, the limit of detection (LOD, 5%-10%) cannot meet clinical testing needs. 5 The throughput and depth of NGS sequencing are significantly improved compared to the first-generation sequencing, and a series of methods for ctDNA detection have been developed.
The method of sequencing
Tagged amplification sequencing (TAM-Seq) is an approach based on NGS after amplifying the target gene with targeted marker primers. This method amplifies the target fragments in advance, thereby solving the problem of the low abundance of the detected targets and improving detection sensitivity. Forshew et al used the TAM-Seq method to detect TP53 mutations in ctDNA in patients with ovarian cancer to achieve early detection of cancer recurrence. This method reduced the detection limit of mutations in ctDNA to 2%, and its specificity and sensitivity were greater than 97%. 27 Cancer personalized profiling by deep sequencing (CAPP-NGS) is a method for individualized analysis through NGS. CAPP-NGS prepares high-frequency mutation libraries through population-level analysis to optimize gene libraries. At the same time, this method designs a “selector” composed of biotinylated DNA oligonucleotides that target the frequent mutations of cancer. The “selector” can be applied to identify cancer-specific genetic abnormalities in patients. 28 Target Error Correct Sequencing (TEC-Seq) is a method that uses NGS to detect low-abundance sequences. This method uses massively parallel sequencing to assess the differences of cfDNA sensitively. The main workflow includes cfDNA and Oligonucleotide Barcode connection, construction of cfDNA library, redundant sequencing, the formation of reference genome queue, and comparison screen out molecular mutations. 29 Cristiano et al 30 tested plasma cfDNA mutations in patients with ovarian cancer and found that the detection rates of cfDNA mutations in patients with stages I to IV were 67%, 75%, 75%, and 81%, respectively, proving that cfDNA mutations are associated with disease progression closely related, it is an available biomarker for early detection of ovarian cancer. Although the sequencing depth of CAPP-NGS is more than 10 000× and the sequencing depth of TEC-Seq is 30 000×, the cost is relatively high, and the data analysis process is more complicated.
The method of PCR
Compared with the sequencing method, the PCR method is simpler and more economical. Quantitative PCR (qPCR), low temperature PCR, digital PCR (dPCR), and droplet digital PCR (ddPCR) are commonly used methods. Martignetti et al 31 used qPCR to detect FGFR2-FAM76A fusion DNA in ctDNA among patients with ovarian cancer to assess tumor recurrence. The results show that ctDNA detection can sensitively find lesions that cannot be detected by CA125, which proves that ctDNA may become a biomarker for the occult period of ovarian cancer. Keserű et al 32 used quantitative real-time PCR to detect the copy number of mitochondrial DNA (mt-DNA) in the whole blood and plasma of patients with serous epithelial ovarian cancer. The results show a significant difference in the mt-DNA copy number of patients’ whole blood compared with healthy volunteers. The mt-DNA copy number of whole blood is closely related to disease progression and increased significantly in the late stage. Low-temperature PCR requires the advanced temperature control ability of the testing equipment, and the LOD is up to 0.05%. Using repressor primers to amplify mutant DNA selectively is a feasible strategy. Chen et al 33 proposed a branch migration-based selective PCR (BM-PCR, Figure 3C) to enrich ctDNA, and this method introduced a blocker. Blocker is matched completely with the wild chain, which can inhibit the extension of the primer to the wild chain and reduce the amplification of the wild chain. Blocker has a base mismatch with the mutant chain, which does not affect the primer work and can enrich low-abundance mutants. However, the LOD is 0.1%, which needs further improvement.
dPCR is a new generation PCR technology used to detect cfDNA/ctDNA. It disperses a sample into a single molecule level, forms multiple copies and distributes them to the microarray panel, and then performs PCR amplification, which is costly. Besides, the technology's early cancer detection rate is still not ideal, less than 50%, limiting its application. 34 Parkinson et al 35 used the dPCR method to analyze the TP53 mutation in ctDNA of ovarian cancer patients. The results showed that the TP53 mutation fragment in ctDNA could be applied to monitor tumor burden (lesion volume > 32 cm3, TP53 mutation amplifiable copy number ≥20/mL); available prediction indicators of progression time (TP53 allelic mutants ≤60% have an estimated sensitivity of 71% [95%CI: 42%-92%] and specificity of 88% [95%CI: 64%-99%]). ddPCR is a method in which samples are dispersed on a microchip in the form of droplets and then amplified. It is an extension of dPCR technology. It takes a short time, and the detection limit can be less than 0.05%. 36 ddPCR method is the most sensitive among PCR, but the detection cost is higher. As a droplet PCR-based technology, BEAMing provides a new method and tool for low-abundance mutation detection. The method is based on 4 parts: bead, emulsion, amplification, and magnetic. 37 BEAMing uses magnetic beads to further enrich the target based on ddPCR, which is often used to detect rare and known mutation sites. Ben et al showed that BEAMing had good consistency with ddPCR methods. 37 Krug et al used BEAMing to detect the epidermal growth factor receptor (EGFR) T790M mutation in ctDNA. 38 The results showed that BEAMing could detect the target mutation at an abundance of 6 copies/mL, demonstrating the excellent sensitivity of the method. However, BEAMing has a high cost and can only detect mutations at known sites, limiting its further application. Vessies et al evaluated the cost of cfDNA detection for 4 methods (ddPCR, Idylla, COBAS z480, and BEAMing), and their results showed that ddPCR and COBAS z480 had the highest detection throughput. In contrast, BEAMing had the highest detection cost. 39
The method of the fluorescent probe
The probe method is a simple and inexpensive method for detecting mutations. However, because traditional probes cannot meet the detection limit and detection throughput requirements, they cannot be applied to detect low-abundance mutations. The detection system of the enzyme-assisted probe can improve the detection sensitivity. Based on the existing BM-PCR, Chen et al used Endonuclease IV (Endo IV)-assisted probe to detect the target mutation chain and reduced the detection limit to 0.01%, which meets the detection requirements of ctDNA. 40 Endo IV can cleave double strands containing gapped bases. As shown in Figure 4A, after being enriched by BM-PCR, the mutant chain is transformed into single-stranded by lambda exonuclease. A fluorescent probe and a blocker add to the above system. The probe has FAM at one end, BHQ at the other, and an empty position in the middle. After adding EndoIV, the probe is cut off, and the fluorophore is released from the quencher to generate a strong signal. The probe designed based on strand migration reaction is simple and highly selective. It can distinguish between mutant and wild-type DNA. Liu et al 41 developed a 5-strand star probe based on the strand migration reaction and combined it with the strand migration reaction (Figure 4B). When the target chain binds to the probe, the chain with the fluorescence quenching group undergoes a chain migration reaction, and the system generates a report signal. This method was applied to detect the plasma BRCA 41293497 mutation in patients with ovarian cancer, and the experimental results proved that the quantitative detection limit of this method was 2 nM. The structure of the target sequence may affect the work of the probe. When the target sequence forms a secondary structure, the probe cannot bind to it and cannot work. Eliminating the secondary structure of the target sequence can improve the working efficiency of the probe. Ming et al 42 introduced 2 additional single-stranded DNAs to the target strand that will form the secondary structure. These 2 single-stranded DNAs match a part of the probe's target region and open the secondary structure formed (Figure 4C). This strategy is applied to detect the plasma BRCA2 rs80359065 mutation in patients with ovarian cancer and reduce the detection limit to 0.05%. 42 The probe designed based on the principle of strand migration reaction is simple and highly selective and can distinguish between mutant and wild-type DNA.

PCR method for evaluating genetic risk of ovarian cancer and related ctDNA detection. (A) The basic workflow of AS-PCR; (B) the universal detection method by the star-probe; (C) the method used by Ming et al to reduce the influence of the secondary structure of the PCR product.
Circular Tumor Cells Detection
Liquid biopsy provides a new method for the early diagnosis of ovarian cancer. In addition to the ctDNA/cfDNA detection described above, circulating tumor cells (CTCs) are another detection target that has attracted much attention. Due to the extremely low abundance of CTCs, pre-enrichment is required before detection. Commonly used enrichment methods are (i) separation based on physical features, such as microfluidic devices; 43 (ii) based on immunoaffinity; the common binding sites are EpCAM, HER2, and MUC1. 44 For the enriched CTCs, the commonly used analytical methods are immunocytochemistry (ICC) and RT-PCR. Nie et al analyzed the diagnostic specificity of HE4 in ovarian cancer-related CTCs using the ICC method, and the results suggested that the specificity of this method was >80%. 45 Chebouti et al used RT-PCR to analyze the expression of EpCAM, ERCC1, MUC1, MUC16, and PI3Kα in CTCs and evaluated the diagnostic specificity of this method. 46 The results suggest that the specificity and sensitivity of this method are >90%. In terms of CTC detection rate, 7.7% to 98% for ICC and 14% to 91% for PCR,46–49 several studies suggest that combined ICC and PCR detection of CTC can effectively improve the detection rate.50,51
CTCs may be closely linked to disease progression and serve as novel targets for assessing prognosis and monitoring treatment for diagnosis significance. Zhang et al's study suggested that the CTC content of samples in advanced disease (Stage III/IV) was significantly higher than that in early disease (Stage I), and similar results were confirmed in other studies. 49 Therefore, CTCs can perform as biomarkers for assessing disease progression. In addition, CTCs can also be used to assess treatment responsiveness. The study by Obermayr et al suggested that the CTC content in samples of platinum-resistant patients was higher than that of platinum-sensitive patients. 52 Poveda et al showed that CTC content decreased in patients after chemotherapy, suggesting that CTC can be a biomarker for assessing chemotherapy response. 53 The sample size of these findings is limited and requires a multicenter, large sample for clinical validation.
Non-Coding RNA Detection
Non-coding RNA (ncRNA) refers to RNA that does not encode a protein, including but not limited to microRNA (miRNA),and long non-coding RNA (lncRNA). The research on miRNA (micro RNA) is the most extensive and in-depth, and miRNA has gradually become an important biomarker for early diagnosis, staging, and recurrence detection of ovarian cancer. Reverse transcription PCR is a common technique for detecting ncRNA. This method identifies a series of diagnostic markers and ncRNAs related to staging and recurrence (Table 3). In addition, simultaneous rapid detection of multiple ncRNAs is crucial. The miRNA microarray constructed by combining target RNA with specific probes can simultaneously detect many miRNAs with strong specificity and high sensitivity. This method has become an essential tool for screening and studying target miRNAs. Zhu et al 64 used miRNA arrays to detect 48 serum miRNAs in patients with epithelial ovarian cancer and patients with benign ovarian tumors. The expression level of 5 miRNAs, including miR-125b, had significant differences. In addition to detecting miRNAs in patients’ blood or tissues through PCR and miRNA microarrays, researchers also need to use a variety of related analysis models to integrate information to more accurately reveal the relationship between miRNAs and the occurrence, development, or recurrence of ovarian cancer. Bagnoli et al 65 set progression-free survival as the clinical observation endpoint. They used semi-supervised prediction methods to predict 35 miRNAs that can predict the progression or recurrence of ovarian cancer. They used the multivariate Cox regression model to establish MiROvaR, a prediction model for ovarian cancer recurrence and evaluated the model's reliability by testing 2 validation sets. Due to the wide variety of available targets of ncRNAs, it is necessary to integrate existing resource information to develop diagnostic and prognostic evaluation tools that can be used in clinical so that ncRNA can play a role in tumor diagnosis and treatment.
The ncRNA Biomarkers for Diagnostic, Progression, and Prognosis Assessment of the Ovarian Cancer.

Abnormal Methylation Detection of Epigenetic Regulation
DNA methylation is one of the earliest discovered and deeply researched epigenetic regulation mechanisms. The state of DNA methylation is closely related to cancer. 66 Abnormal methylation is associated with early cancer. Therefore, the state of DNA methylation can be regarded as a biomarker for cancer diagnosis and prognosis (Table 4). Currently, the typical analysis methods include: pyrosequencing, methylation-specific restriction endonuclease analysis, quantitative methylation-specific PCR, and methylation-specific high resolution melting analysis.66–70 These methods use sulfite to transform DNA and then read and analyze sequence information through PCR amplification and sequencing. The cytosine in the sulfite-treated DNA will be converted into uracil, while the methylated cytosine will not change (Figure 5). By measuring the ratio of cytosines, the degree of methylation can be assessed.
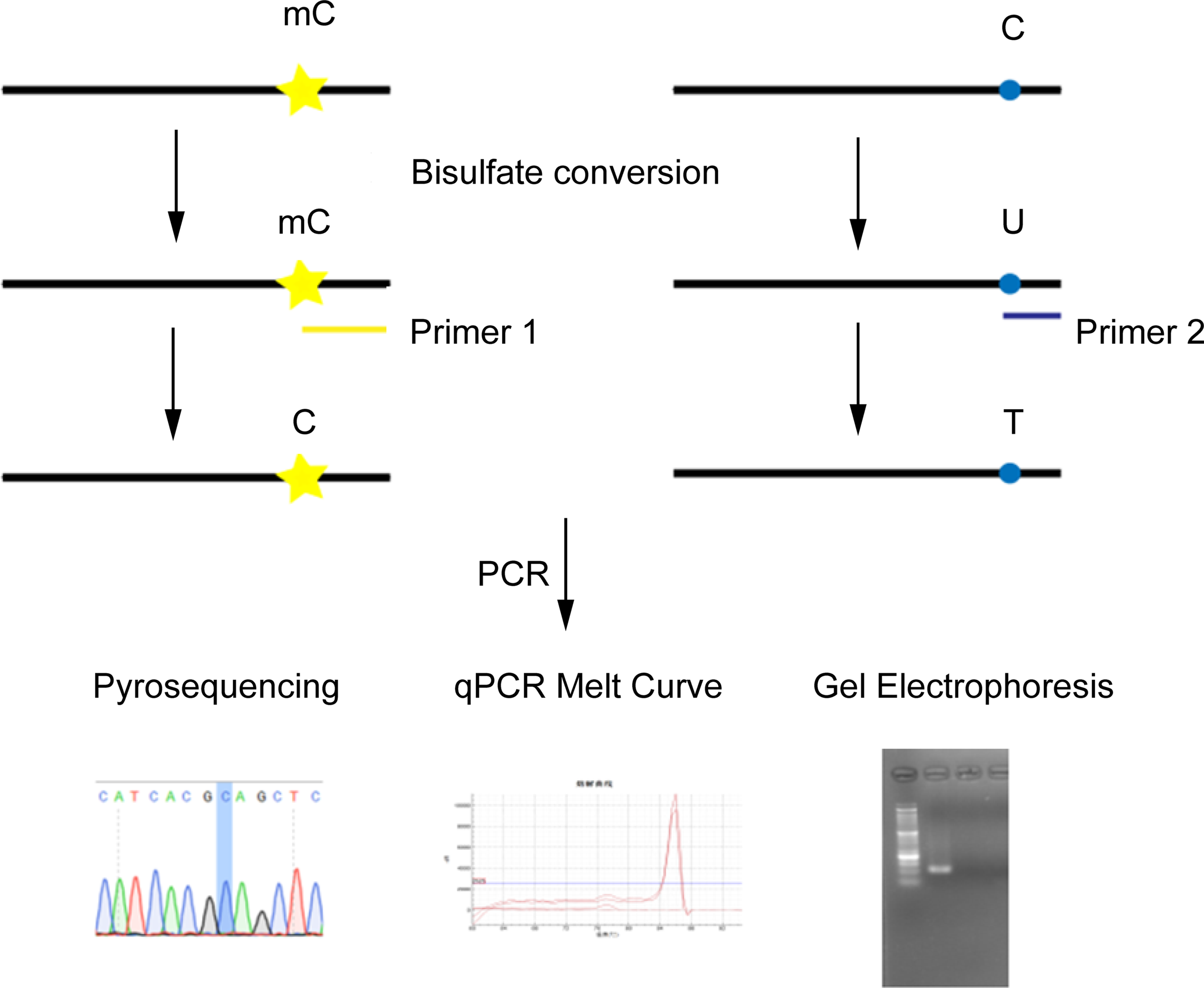
The basic workflow for DNA methylation analysis.
The Abnormal Methylation for Diagnostic, Progression, and Prognosis Assessment of the Ovarian Cancer.

In addition to DNA methylation, RNA methylation is also regarded as another valuable biomarker for evaluating tumor occurrence, development, and prognosis. 77 RNA methylation is the main form of RNA modification, including N6-methyladenine (m6A), N7-methylguanine (m7G), and 5-methylcytosine (m5C), N-methyladenine (m1A). m6A is one of the most common and abundant internal modifiers of RNA molecules,77,78 which determines the expression or inactivation of specific genes to a certain extent and is related to cell growth, development, death, and malignant transformation. Li et al 79 found that after miRNA-9 was methylated, ovarian epithelial cancer cells were resistant to paclitaxel by targeting cyclin G1 (CCNG1). Mass spectrometry, colorimetry, PCR, sequencing, and PCR and sequencing are commonly used to evaluate RNA methylation levels. We take m6A detection as an example. Gene-specific m6A-qPCR, m6A sequencing (m6A-seq), and cross-linking and immunoprecipitation sequencing (CILP-seq) are widely used methods. All of the above techniques require m6A-specific antibodies to enrich the target. The m6A-qPCR method (Figure 6) is based on RNA fragmentation, using specific antibodies to identify the target site and complete immunoprecipitation to enrich the RNA. After eluting the target RNA, the level of m6A is shown by qPCR analysis. 80 The m6A sequencing method first collects specific antibody-enriched fragments and then constructs a high-throughput library for sequencing. This method requires the construction of a transcriptome library as a negative control at the same time. After comparing the 2, the regions with high methylation levels can be found by bioinformatics analysis. 81 This method can perform a high-throughput and quantitative assessment of the RNA methylation level in the corresponding region. However, it cannot distinguish whether a single base is methylated or not, and its accuracy is insufficient. The CLIP-seq method makes up for this shortcoming. This method uses specific antibodies to label the target fragments and then ultraviolet light to cross-link the RNA for reverse transcription. In reverse transcription, the labeled target site is truncated and mutated, and the detection of single base site m6A is completed by subsequent high-throughput sequencing. 82 Liu et al 83 showed that the m6A modification of EIF3C mRNA promotes protein translation of EIF3C, thereby promoting the occurrence and metastasis of ovarian cancer.

The main workflow for detection of m6A.
The epigenetic modification of m6A is also closely related to the invasion and metastasis of ovarian cancer. METTL3 is one of the RNA N6-methyladenosine methyltransferases. Hua et al 84 found that the highly expressed METTL3 in ovarian cancer cells promotes the m6A modification of the receptor tyrosine kinase AXL, thereby up-regulating its expression level and promoting epithelial-mesenchymal transition, which contributes to the growth and invasion of ovarian cancer. Also, m6A modification is closely related to chemotherapy resistance of ovarian cancer. Research by Fukumoto et al 85 showed that m6A modification of FZD10 mRNA improves its stability and makes the expression of FZD10 up-regulation. Overexpression of FZD10 can up-regulate the resistance of epithelial ovarian cancer cells with BRCA gene mutations to PARPis through the Wnt/β-catenin pathway. RNA methylation has become a research hotspot in tumor epigenetic modification, and it is reasonable to believe that it can be a valuable indicator for evaluating cancer occurrence and prognosis.
Extracellular Vesicles Detection
Extracellular vesicles are a general term for lipid bilayer-enclosed vesicles secreted by cells to the outside, including exosomes, microvesicles, oncosomes, and microparticles. 86 Extracellular vesicles play an essential role in the interaction of information between cells. Recent studies have suggested that EVs are closely related to the tumor microenvironment and biological processes such as tumorigenesis and metastasis.87–90 The coated tumor-related nucleic acid molecules provide a new target for gene diagnosis of tumors and have become a hot spot in oncology research. miRNAs within vesicles and proteins located on the surface or inside vesicles are essential targets in related research.
EV-related miRNAs are emerging targets for ovarian cancer liquid biopsy, and the commonly used detection method is qRT-PCR. Zhu et al analyzed miRNA expression in serum exosomes from 36 patients with malignant ovarian tumors, 31 patients with benign ovarian tumors, and 32 healthy volunteers. The results showed that miR-205 was related to the occurrence and metastasis of ovarian cancer. The results demonstrate that it can be used as a target for early screening and prognostic assessment. 91 Similarly, Liu et al suggest that miR-4732-5p may be an early diagnostic marker for EOC. 92 Besides serving as diagnostic markers, EV-associated miRNAs and mRNAs are also closely related to diagnosis and prognostic assessment. Some results showed that EV-associated miRNAs are associated with the transformation of cancer-associated fibroblasts and promote the formation of the tumor microenvironment. 93 Zhou et al proved that the exosomal miRNAs from EOC-associated macrophages are related to immunosuppression. Five EV-associated miRNAs can induce Treg cell differentiation, inhibiting tumor immunity. 94 We present the related miRNA information in Supplemental Table S1.
In addition to miRNAs, proteins on vesicle membranes are also essential biomarkers for the early screening of ovarian cancer. Common ovarian cancer markers CA125 and HE4 were also detected in exosomes. The specificity and sensitivity of CA125 and HE4 in serum exosomes for early diagnosis of ovarian cancer were 83.3% and 96.9%. 91 In addition, Liang et al proved that the protein characteristics of EOC-derived exosomes could help diagnose ovarian cancer. Moreover, the related proteins include EGFR, epithelial cell adhesion molecule (EpCAM), and ERBB (see Supplemental Table S2 for details). 95 The proteins in these ovarian cancer-related microvesicles have been reported many times. Reiner et al analyzed EV-related proteins in ascites of high-grade serous ovarian cancer patients, and the results suggested that matrix metalloproteinase 9 (MM9) can be used as a specific marker for high-grade serous ovarian cancer. 96 The related EV-related proteins are involved in important biological processes such as ovarian cancer metastasis and the information interaction of the tumor microenvironment. The related targets are summarized in Supplemental Table S2.
Image Related Genetic Detection
Ultrasound, CT, and magnetic resonance imaging are commonly used imaging methods for ovarian cancer screening. Although ultrasonography is recommended as the first-line imaging modality, there are still approximately 20% to 25% of cases where the diagnosis cannot be made when evaluating the malignant potential of ovarian masses. 12 MRI can help identify the above indeterminate cases. The ADNEX MR scoring system has a specificity of 96.6% and a sensitivity of 93.5% for judging benign and malignant pelvic masses. 9 CT is the preferred method for staging ovarian cancer. CT is used to detect residual tumor lesions after surgery, which is a crucial method for evaluating the prognosis of patients. When CT combines with PET, tumor recurrence can be effectively assessed.
In recent years, new nucleic acid nano-probes or nanomaterials based on CT and MRI have provided new methods and strategies for molecular imaging. Molecular imaging of KRAS, BRAF, TP53, and other mutation sites provides auxiliary information for distinguishing indolent ovarian cancer from invasive ovarian cancer. 97 MRI molecular imaging based on magnetic materials provides a new strategy for related research. Zhao et al constructed an MRI-based molecular imaging method of MUC1 using magnetic iron oxide nanomaterials combined with MUC1-specific peptides. 98 The Fe3O4 magnetic nanomaterial constructed by Zhang et al realized MRI-based siBIRC5 imaging. 99 In addition, molecular imaging methods based on PET/CT or SPECT/CT provide new methods for assessing tumor aggressiveness. Biabani et al used a 68Ga-PET tracer to achieve HER2 molecular imaging of ovarian cancer cells. 100 Similarly, Radioiodinated 4-(p-Iodophenyl) Butanoic Acid-Modified Estradiol Derivative achieved SPECT/CT-based molecular imaging of ER. 101 The above new methods fully expand the application space of tumor imaging and provide new tools for precision medicine.
Discussion and Conclusion
The occurrence and development of tumors are closely related to genes. In the era of precision medicine, detecting and evaluating cancer at the genetic level is the only way to achieve precise diagnosis and individualized diagnosis and treatment. Researchers have developed tools for detecting tumor-related gene mutations based on sequencing, PCR, and probes, and are trying to reduce costs, lower detection limits, and increase throughput to achieve truly specific, sensitive, and economical detection. From tissue to liquid biopsy, tumor gene detection technology has made a great leap toward non-invasive, specific, and highly sensitive.
With the continuous expansion of essential medicine and clinical medicine knowledge, cancer-related gene mutations, ncRNA expression abnormalities, and epigenetic modifications have provided a large amount of available genetic information for early cancer diagnosis, disease staging, and prognostic evaluation providing genetic diagnosis, feasible methods and strategies. However, we must also clearly see that there is still no genetic diagnosis tool available for clinical use of cancer, and clinical diagnosis cannot be made based on genetic test results alone. Therefore, the cancer-associated genetic diagnosis still needs further development. In the field of genetic testing for ovarian cancer, the author believes that it can be developed from the following aspects.
First, study the internal mechanism of the occurrence and development of cancer. Research the causes and mechanisms of ovarian cancer formation, metastasis and invasion, loss of the original programmed death function of cancer cells, immune response and monitoring failure, and fully explore the available information. In recent years, research related to metabolism, tumor microenvironment, gut microbes, and immunity has become hotspots in tumor research. Newer cell death mechanisms have opened new horizons, such as cuproptosis and ferroptosis. There are some new methods for finding new targets, such as metabolomic analysis, single-cell sequencing, and spatial transcriptomic analysis. They provide a powerful tool to explore new mechanisms of tumorigenesis.
Second, develop specific and sensitive detection tools. On the one hand, specifically distinguishing cancer tissues/cells and normal tissues/cells is an essential requirement for cancer detection tools. Insufficient specificity may lead to false-positive results and produce a wrong diagnosis and treatment plans. On the other hand, low-abundance mutation detection is one of the essential methods for the early detection of cancer, which puts forward specific requirements on the sensitivity of the detection method. However, the LOD for the methods cannot meet the clinical requirements, which often leads to false-negative results. It can even delay the patient's optimal treatment time or make the wrong choice of a treatment plan. Besides, the existing methods that reach the detection limit often have complex procedures and high costs. It is hard to promote these methods. It is necessary to combine multiple disciplines, sequencing, PCR, probes, and other methods to develop specific, sensitive, and economically efficient detection tools. For example, the probe with the CRISPR-Cas system demonstrates superior ability in detecting SARS-Cov-2, and the LOD can reach 0.001%, which provides a new method for rapid detection of low-abundance mutations. 102
Third, develop new tools and platforms for processing complex genetic information. There are many biomarkers related to ovarian cancer genetic diagnosis (cancer-related genes, cancer-related transcription factors, ncRNA, methylation, etc) However, it is difficult to effectively and fully utilize them. Therefore, it is necessary to develop effective analysis tools to integrate information. For cancer diagnosis and treatment, synthetic biology can build an automated genetic information integration platform. This method programs target cells with artificial gene circuits so that they can respond to biomarkers in the environment and perform predetermined functions. 103 The AND gate circuit constructed by combining multiple cancer biomarkers can specifically distinguish cancer cells from normal cells. This method has been used in the diagnosis and treatment of a series of cancers such as bladder cancer 104 and liver cancer. 105 The effective implementation of this method reminds us that we need to fully apply existing targets to construct a strategy that can be transformed from the laboratory to the clinic.
Genetic testing is a useful tool for early diagnosis and prognostic evaluation of ovarian cancer and has broad application prospects. We believe that individualized genetic diagnosis and disease assessment will be applied widely in the future.
Supplemental Material
sj-docx-1-tct-10.1177_15330338221114497 - Supplemental material for The Progress of the Specific and Rapid Genetic Detection Methods for Ovarian Cancer Diagnosis and Treatment
Supplemental material, sj-docx-1-tct-10.1177_15330338221114497 for The Progress of the Specific and Rapid Genetic Detection Methods for Ovarian Cancer Diagnosis and Treatment by Kejun Dong, Wei Zhang, Shuangshuang Cheng, Wan Shu, Rong Zhao and Hongbo Wang in Technology in Cancer Research & Treatment
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by the National Key R&D Program of China, (Grant Nos. 2018YFC0114600, 2018YFC0114605), the Science and Technology Innovation Project of Hubei Province (No. 2019ACA138), Wuhan Science and Technology Bureau of Hubei Province of China (2019020701011430), State Key Laboratory of Analytical Chemistry for Life Science (SKLACLS2105), 2020 Graduate Teaching Reform Research Project of HUST (YY202029).
Supplemental Material
Supplemental material for this article is available online.
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.