
Abstract
Introduction
Nasopharyngeal carcinoma (NPC) is a malignant tumor in the head and neck that is prevalent in South China, especially in Guangdong Province and the Guangxi Zhuang Autonomous Region. 1 In the past few decades, although substantial improvements have been made in radiotherapy and chemotherapy,2,3 the clinical outcomes of NPC patients are still not satisfactory. Distant metastasis is the leading cause of treatment failure in NPC patients, and approximately 30% to 40% of locoregionally advanced NPC patients eventually develop distant metastasis after radical treatment. 4 At present, the main therapeutic strategy used to reduce the risk of distant metastasis in clinical practice is to treat patients at high risk of distant metastasis with systemic chemotherapy.5–7 Although the risk of NPC patients developing distant metastasis can be partly characterized by TNM staging, the prediction accuracy is only 63.7%. 7 For instance, distant metastasis still occurs in 13% of stage N1 patients with low risk, but 65% of stage N3 patients with high risk do not develop distant metastasis. 8 Therefore, TNM staging is not precise enough to predict the risk of distant metastasis after radical therapy, and a more accurate method is warranted to determine the trend of distant metastasis after treatment and improve treatment strategies.
It is well known that the distant metastasis of cancer is an extremely complicated multistep process that consists of a succession of cell biological changes termed the invasion–metastasis cascade.9,10 The mechanism underlying these biological changes is the high instability of the cancer genome (eg base deletions and insertions, gene amplifications, epigenetic dysregulation, and posttranscriptional and posttranslational modifications). Because mRNA expression profiles may be determined by multiple changes above, we performed high-throughput screening of mRNA profiles in 92 tissue samples of NPC with the goal of screening out the differentially expressed genes (DEGs) between NPC patients with and without distant metastasis. Subsequently, gene functional enrichment analysis was performed to elucidate and visualize the molecular mechanisms underlying the distant metastasis process of NPC. Then, through feature selection performed using least absolute shrinkage and selection operator (LASSO)-penalized Cox regression, four candidate genes were selected. Based on the regression coefficients of these 4 genes, a risk prediction model was established. This model was tested by performing survival analysis in high-risk and low-risk samples from both the training and validation cohorts, and the performance of the model was evaluated using the time-dependent receiver operator characteristic (ROC) curve. To verify independence of prognostic effect of gene model and other clinical features, we further integrate patients of 2 validation cohorts (Beijing validation cohort and Wuzhou validation cohort) to perform univariate and multivariate analysis using Cox proportion hazard model.
Comparison of mRNA expression profiles between cancer cells and normal control cells or between cancer cells at different developmental stages or disease states could promote the discovery of characteristic gene signatures associated with oncogenesis and cancer progression. Recently, it has been suggested that certain gene expression patterns can be used as molecular models for early diagnosis, subgroup classification and even for risk prediction of distant metastasis in patients with locoregionally advanced NPC.11–14 To this end, we performed this study with the aim of exploring the potential molecular mechanisms underlying the distant metastasis process of NPC and establishing a 4-gene model that can evaluate the risk of developing distant metastases and is suitable for patients with all stages of NPC.
Materials and Methods
Patient Selection and Tissue Sample Collection
A total of 92 patients were newly diagnosed with NPC between September 2007 and September 2012. Forty-four of the 92 patients were from People's Hospital of Guangxi Zhuang Autonomous Region, 25 of the 92 patients were from Xuanwu Hospital of Capital Medical University, and 23 of the 92 patents were from Wuzhou Red Cross Hospital. The present study was approved by the ethical committee of People's Hospital of Guangxi Zhuang Autonomous Region (No: KY-2019-001), Xuanwu Hospital (CLE2021-153), and Wuzhou Red Cross Hospital (S2019-35). All patients signed informed consent documents prior to participating in this study. The identity details of all patients enrolled in this study have been de-identified. The eligibility criteria of patients for inclusion in the study were as follows: (1) pathological confirmation of undifferentiated nonkeratinized carcinoma of the nasopharynx; (2) Union for International Cancer Control (UICC) staging system 2010 clinical classification of I to IVb, without a history of other malignant tumors or anticancer therapy; and (3) Karnofsky performance score ≥70. The exclusion criteria included a history of severe systemic diseases, pregnancy or lactation, and the presence of a contradiction for receiving chemotherapy, radiotherapy, or surgery. Fresh NPC tissue samples of 92 NPC patients were obtained by nasopharynx biopsy under narrow-band imaging (NBI) endoscopy prior to anticancer therapy and then frozen with liquid nitrogen (−196 °C) until analysis.
Pretreatment Evaluation of Patients
All of the patients underwent a pretreatment evaluation that included a precise clinical examination of the head and neck region, fiber optic nasopharyngoscopy, head and neck magnetic resonance imaging (MRI), chest x-ray, ultrasonography of the abdominal region, bone scan, and a complete blood count and biochemical profile.
Patient Treatment
All patients were treated with intensity-modulated radiotherapy (IMRT) once a day 5 times a week. The target area was delineated based on the tumor boundary shown in MRI and computed tomography (CT). The prescribed doses were 69.76 to 76.3 Gy for PGTVnx, 64 to 70 Gy for PGTVnd, 59.4 to 64.0 Gy for PTV1, and 50.0 to 54.0 Gy for PTV2. All patients were administrated with concurrent chemotherapy including cisplatin (40 mg/m2) or carboplatin (500 mg/m2), and fluorouracil (2000 mg/m2) with intervals of 21 days between cycles.
Patient Follow-up
All the patients who had completed the NPC treatment attended follow-up visits every 3 months for the first year, every 6 months for the second to the fourth years, and annually thereafter. The follow-up methods were as follows: (1) All patients received Epstein–Barr (EB) virus serological examination and nasopharyngeal endoscopic examination, and endoscopically positive patients underwent nasopharyngeal biopsy. (2) All patients were confirmed by nasopharyngeal skull base MRI, chest x-ray, and cervical lymph node and abdominal ultrasonic examinations. (3) Patients who had been diagnosed with abnormal bone metabolism by emission computed tomography (ECT) scans received positron emission tomography (PET)/CT scans. The survival time was calculated from the date of first treatment completion to either the date of an event of interest (death caused by NPC or the development of distant metastasis) or the end of the follow-up.
Procedure
Tissue RNA was isolated from fresh frozen tissue samples using the QIAGEN AllPrep DNA/RNA Mini Kit (Qiagen, Cat# 80204) according to the manufacturer's protocol. RNA degradation and contamination were monitored on 1% agarose gels. RNA purity was checked using a NanoPhotometer® spectrophotometer (Implen) or Nanodrop 2000 (Thermo Scientific). RNA concentration was measured using the Qubit® RNA Assay Kit on a Qubit®2.0 Fluorometer (Life Technologies). RNA integrity was assessed using the RNA Nano 6000 Assay Kit on an Agilent Bioanalyzer 2100 system (Agilent Technologies).
For high-quality samples, a total amount of 1.5 μg RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using the NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer's recommendations, and index codes were added to attribute sequences to each sample. In brief, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5X). First-strand cDNA was synthesized using random hexamer primers and M-MuLV Reverse Transcriptase (RNaseH-). Second-strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. The remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of the 3′ ends of DNA fragments, NEBNext adaptors with hairpin loop structures were ligated to prepare for hybridization. To select cDNA fragments of correct length, the library fragments were purified with the AMPure XP system (Beckman Coulter). Then, 3 μl of USER Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37 °C for 15 min followed by 5 min at 95 °C before PCR. PCR was performed with Phusion High-Fidelity DNA polymerase, universal PCR primers, and Index (X) Primer. Finally, the products were purified (AMPure XP system), and library quality was assessed on an Agilent BioAnalyzer 2100 system (Agilent Technologies). For low-quantity RNA samples (typically less than 100 ng of total RNA), the NEB Next rRNA Depletion Kit (Human/Mouse/Rat) (NEB E6310, USA) was used to deplete rRNA. Next, cDNA was synthesized from rRNA-depleted total RNA with the NEBNext RNA First-Strand Synthesis Module (NEB E7525S, USA), and double-stranded cDNA was generated from first-strand cDNA with the NEB Next mRNA Second-Strand Synthesis Module (NEB E6111S, USA) according to the manufacturer's instructions. DNA fragmentation and adapter ligation were carried out using the TruePrepTM DNA Library Prep Kit V2 for Illumina (Vazyme, China). The concentration of each library was quantified by the Qubit® RNA Assay Kit on a Qubit® 2.0 Fluorometer (Life Technologies) according to the manufacturer's instructions. The size distribution was evaluated using an Agilent high-sensitivity chip on an Agilent Bioanalyzer 2100 system (Agilent Technologies).
For mRNA sequencing, clustering of the index-coded samples was performed on a cBot Cluster Generation System using the HiSeq 4000 PE Cluster Kit (Illumina) or NextSeq 500/550 High Output Kit V2 (Illumina) according to the manufacturer's instructions. After cluster generation, the library preparations were sequenced on an Illumina HiSeq 4000 or NextSeq 500 platform, and 150 bp paired-end reads were generated.
For data processing, Cutadapt was first used to trim adapter contamination and error-prone low-quality bases with the following command line parameters: --quality-base = 33 --quality-cutoff = 20,20 --format = fastq -a CTGTCTCTTATACACATCT -A CTGTCTCTTATACACATCT -g AGATGTGTATAAGAGACAG -G AGATGTGTATAAGAGACAG --times = 6 --minimum-length = 20 --max-n = 0.1 --trim-n. Bowtie2 was used with default parameters to remove rRNA-containing reads by mapping reads to 5S rRNA, 5.8S rRNA, 18S rRNA, and 28S rRNA sequences, as well as mitochondrial 16S rRNA and 12S rRNA sequences. High-quality reads were then aligned to the GRCh38 reference genome using STAR with the following parameters: --outSAMunmapped Within --outFilterType BySJout --outFilterMultimapNmax 20 --outFilterMismatchNoverLmax 0.04 --alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 1000000 --alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --sjdbScore 1 --runThreadN 2 --genomeLoad NoSharedMemory --outSAMtype BAM Unsorted --quantMode TranscriptomeSAM --readFilesCommand zcat. Normalized gene expression (fragments per kilobase million [FPKM] and transcripts per kilobase million [TPM]) was calculated by RSEM with the following parameters: -p 4 --seed-length 20 --paired-end --bam --estimate-rspd.
Statistical Analysis
Calculation and justification of the sample size selected for this study
To determine the sample size required for an RNA sequencing (RNA-Seq) experiment to identify differentially expressed genes (ie prognostic genes) between metastasis-positive group and metastasis-negative group, we used the R package RnaSeqSampleSize to calculated the minimum sample size. We assume that the minimum average read counts among the prognostic genes in the control group is 20, the dispersion for each gene is 0.1, and false discovery rate (FDR) threshold is set to be 0.2. We also suppose that the total number of genes for testing is 20,000 and the proportion of non-differentially expressed genes is 90%, and the minimum fold change is 1.5. By calculation, we will need to study 16 subjects in each group to be able to get an average power of 0.8.
Identification of DEGs
We select 16 pairs NPC tissues from 32 patients with or without distant metastasis after curative treatment. These 32 patients were selected from the total 92 patients enrolled in this study and were strictly matched by sex, age, TN stage, and therapeutic strategies to rule out the influence of these confounding factors on distant metastasis. Genes that were either not expressed or expressed at low levels (<5 reads in half of the samples) were removed prior to differential expression analysis. DESeq2 is a popular tool for the identification of DEGs. Since it requires raw counts for each gene as input, HTSeq was used to count reads mapping to each gene. The gene expression profiles of patients who developed metastasis within 5 years after diagnosis were compared to those of patients who remained metastasis-free for at least 5 years. Genes that met the following criteria were identified as DEGs: (1) fold change >1.5 or fold change < 0.667 and (2) adjusted P value <.2.
Feature selection, risk model construction in the training cohort, and risk model evaluation in the validation cohort
First, 48% (44 out of 92) of the RNA-Seq samples from People's Hospital of Guangxi Zhuang Autonomous Region were selected as the training cohort with the remaining samples from Xuanwu Hospital of Capital Medical University and Wuzhou Red Cross Hospital were selected as the validation cohorts. Training samples were selected to ensure that the distribution of the selected samples was consistent with all samples in terms of age, sex, and clinical stage. After DEGs were identified from the training cohort, LASSO was applied to select prognostic genes. LASSO constructed a penalty function to obtain a more refined model so that it compressed some coefficients, and at the same time, it set some coefficients to zero. LASSO-based feature selection was conducted using the glmnet package in R software (version 3.4.0). The tuning parameter lambda was determined according to the expected generalization error estimated from 10-fold cross-validation, and we adopted the lambda value that gave minimum mean cross-validated error, known as lambda.min. A Cox proportional hazards regression risk model was then constructed to predict prognosis with genes selected by the LASSO algorithm. The risk scores of each sample from the training and validation cohorts were calculated according to the risk model. The respective medians of the 3 cohorts were used as the cut-off value to divide patients into high-risk and low-risk groups. Kaplan–Meier curves were plotted to compare the distant metastasis-free survival (DMFS) of high-risk and low-risk patients. P values and hazard ratios (HRs) with 95% confidence intervals (CIs) were generated by the log-rank test. The time-dependent ROC curve was plotted with the time ROC R package to characterize the discrimination potential of the risk score. The area under the ROC curve (AUC) was calculated as well. The univariate and multivariate analyses were performed using the Cox proportion hazard model.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) (version 3.0, http://software.broadinstitute.org/gsea/downloads.jsp) was performed between patients with and without metastasis within 5 years. 14 The Signal2Noise metric, which calculates the difference of means scaled by the standard deviation, was used to rank the genes. The ranked genes were compared against curated gene sets in the Molecular Signatures Database (http://software.broadinstitute.org/gsea/msigdb) for enrichment analysis. The meandiv normalization method was used to obtain the enrichment scores of the gene sets. A total of 1000 permutations were performed to generate a null distribution for the enrichment scores of the hallmark gene sets and functional annotation gene sets. The gene sets used for enrichment analysis included “h.all.v6.2.symbols.gmt” and “c2.cp.kegg.v6.2.symbols.gmt”. A cut-off of nominal P ≤ .05 was applied to identify significantly enriched gene sets.
Results
Patient Clinical Characteristics and Follow-up Outcome
latest patient follow-up visit occurred in July 2017. The follow-up time ranged from 2 to 114 months, with a median of 64 months. The flow chart of this study is shown in Figure 1. Among all 92 patients enrolled in this study, 44 patients from People's Hospital of Guangxi Zhuang Autonomous Region were included in the training cohort, 25 patients from Xuanwu Hospital of Capital Medical University were included in the Beijing validation cohort, and 23 patients from Wuzhou Red Cross Hospital were included in the Wuzhou validation cohort. All patients achieved clinical cure after the completion of the first treatment. Thirty-one of the 92 patients eventually developed distant metastasis, and 61 of the 92 patients did not develop distant metastasis by the end of the follow-up. The clinical characteristics of the 92 patients are summarized in Table 1.
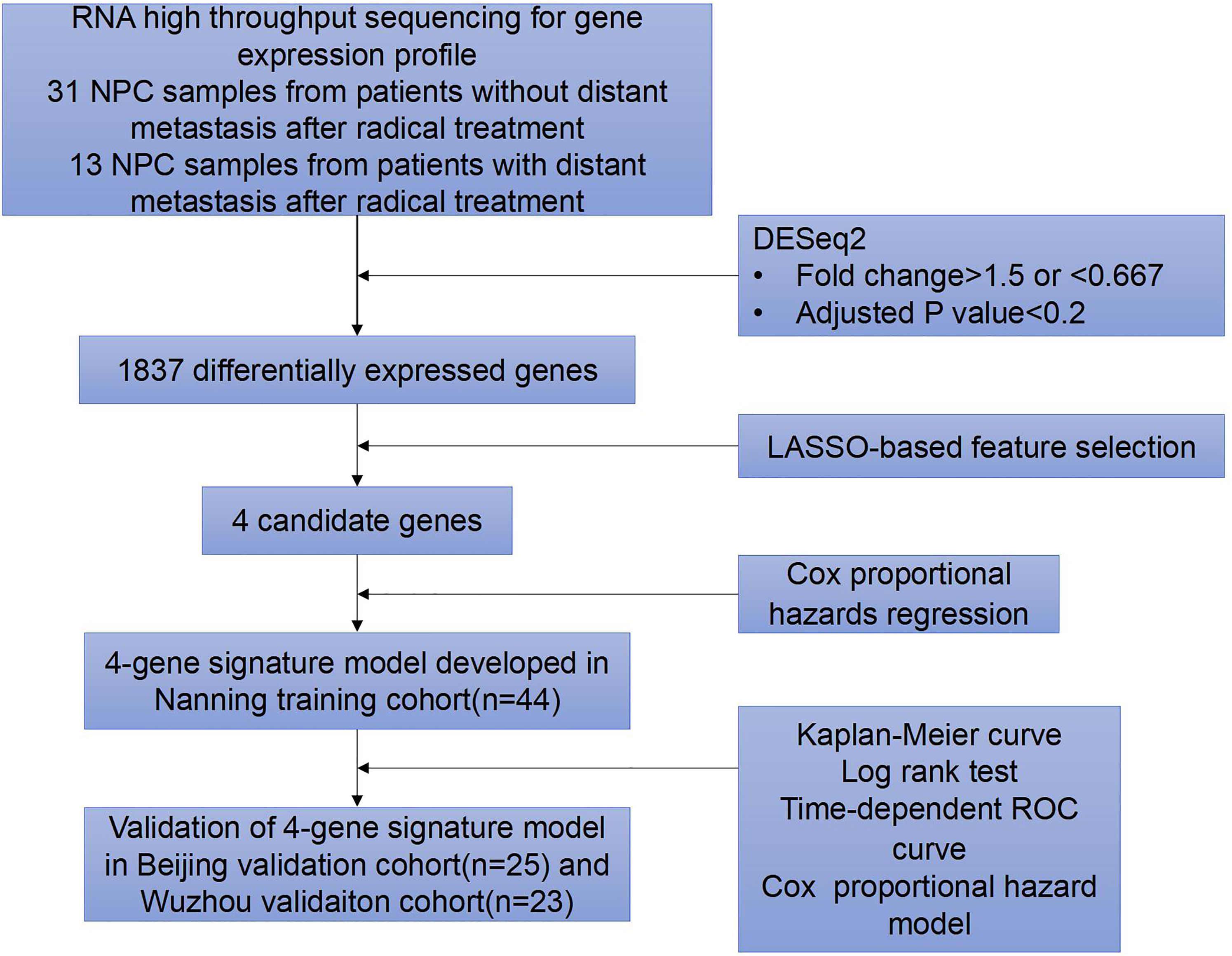
Flow chart of the study.
The Clinical Characteristic of the 92 NPC Patients.
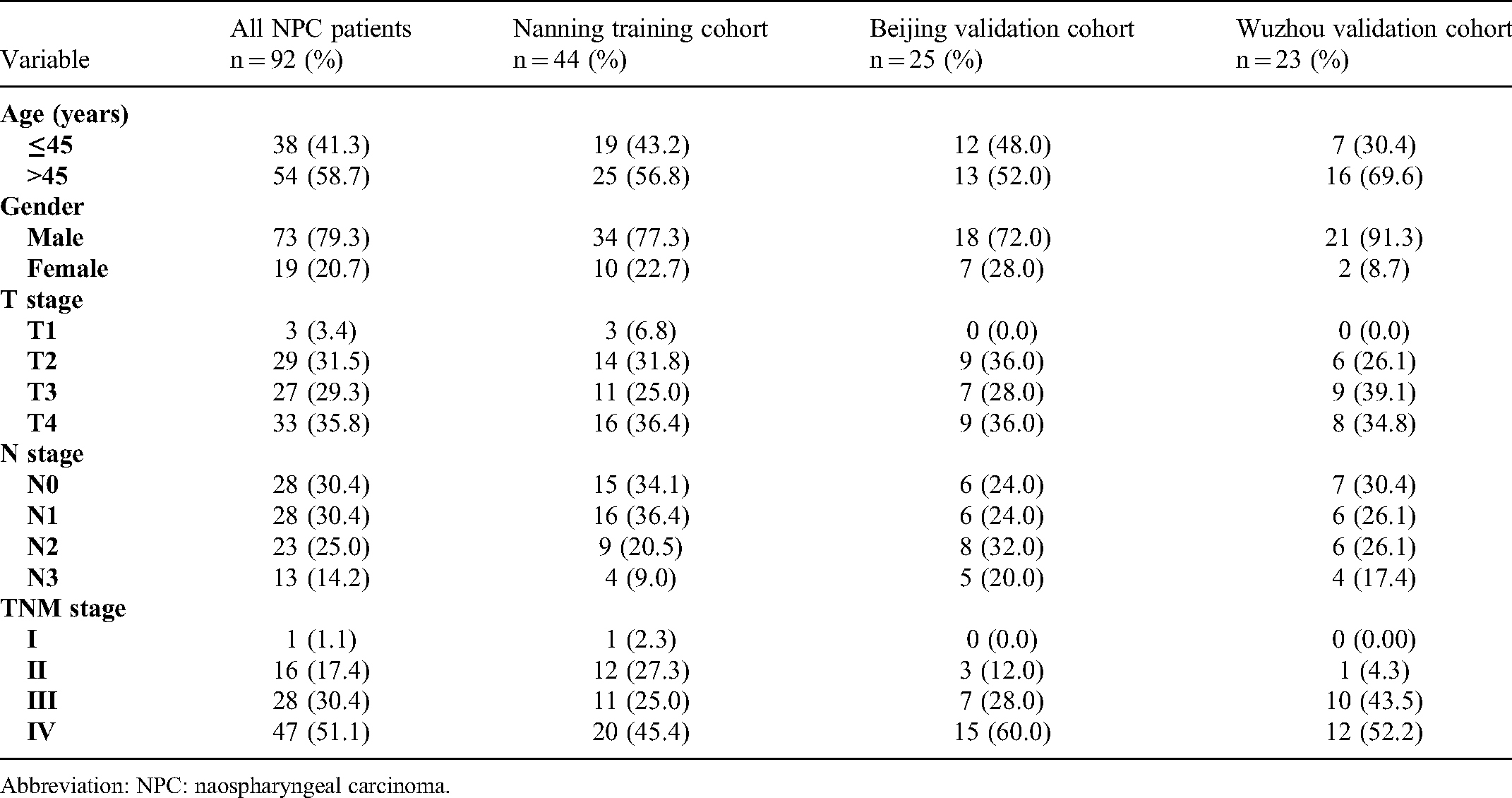
Abbreviation: NPC: naospharyngeal carcinoma.
DEG Screening and GSEA
In the bioinformatics analysis, 1837 mRNAs, including 869 upregulated genes and 968 downregulated genes, were found to be differentially expressed between 22 patients with nonmetastatic NPC and 47 patients with NPC who developed distant metastasis after treatment. The volcano plot and heatmap of DEGs are shown in Figure 2A and B. GSEA under the cut-off criteria defined by nominal P value<.05 identified 6 gene sets, including the Wnt/β catenin signaling pathway, hedgehog (Hh) signaling pathway, Notch signaling pathway, mitotic spindle, apical surface, and estrogen response late, which were enriched in patients with distant metastasis (Figure 3).
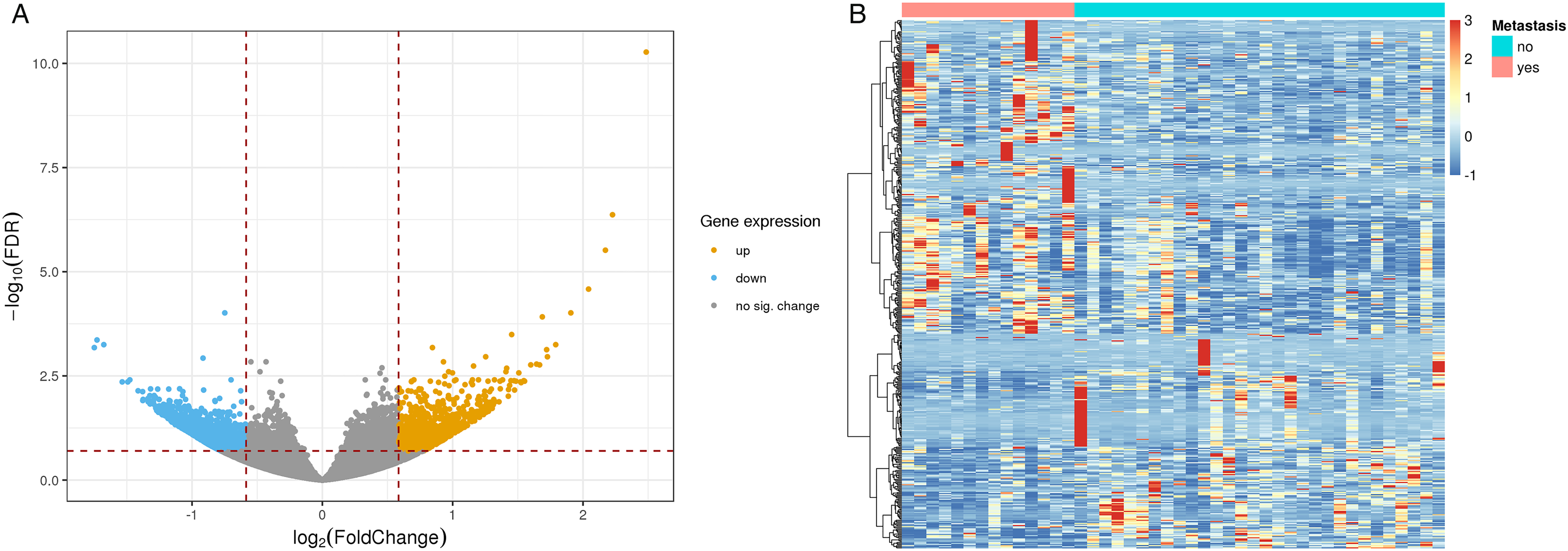
Differentially expressed genes in patients with and without distant metastasis. (A) Volcano plot of 1837 differentially expressed genes that distinguish patients with and without distant metastasis. (B) Heatmap of 1837 differentially expressed genes that distinguish patients with and without distant metastasis. Patients are in columns and genes are in rows.
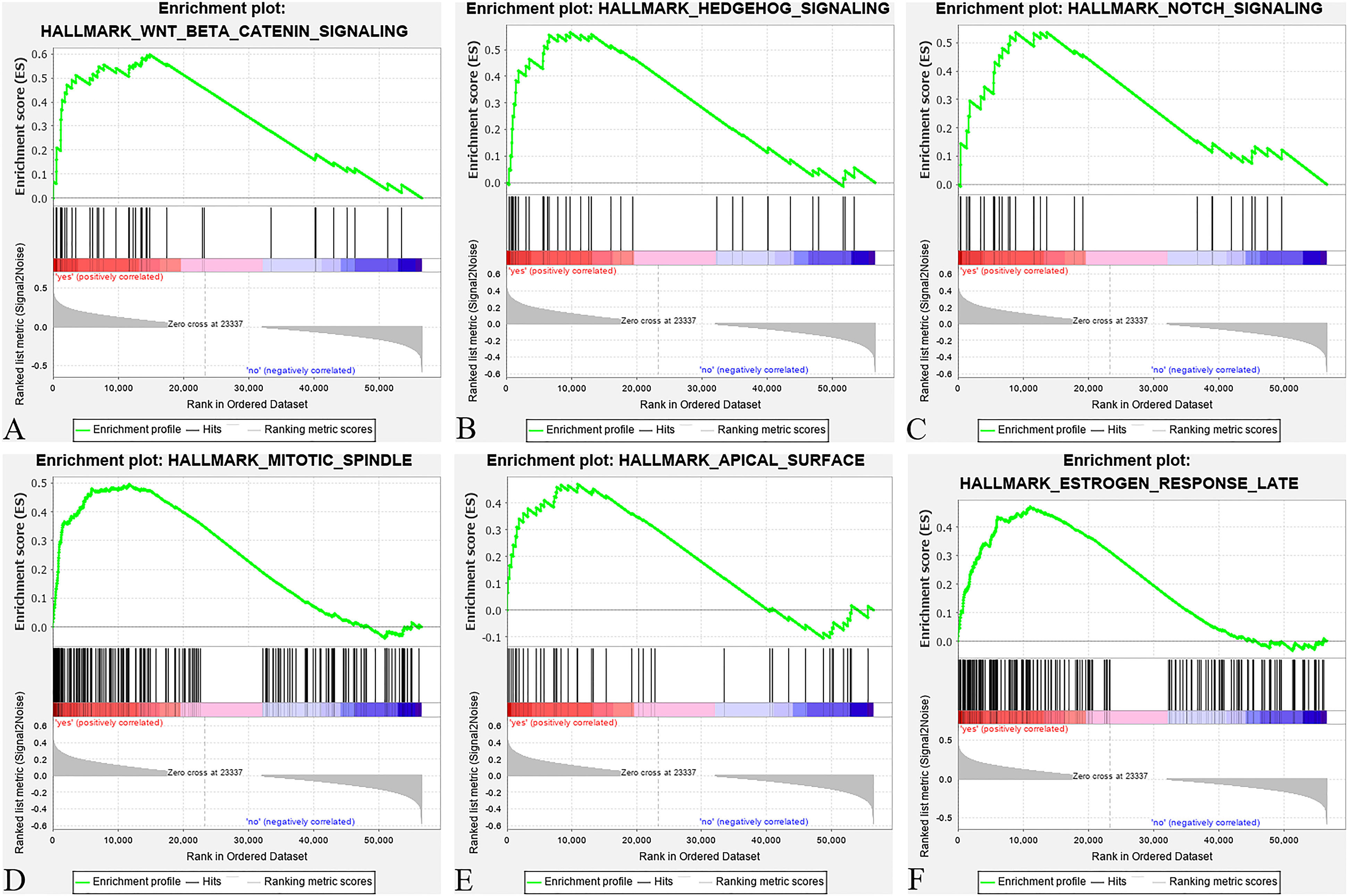
Gene set enrichment analysis (GSEA) of gene expression data in nasopharyngeal carcinoma (NPC). Six gene sets, including Wnt β catenin signaling pathway (A), hedgehog signaling pathway (B), Notch signaling pathway (C), mitotic spindle (D), apical surface (E), and estrogen response late (F), were enriched in the patients with distant metastasis.
Construction of a 4-Gene Signature Model in the Training Cohort
Through feature selection performed by LASSO Cox regression, a four-gene signature model was identified based on the optimal value of lambda (Figure 4A and B). The risk score was calculated for each patient using the following formula derived from the expression level of these four genes weighted by their corresponding regression coefficient: Risk score = (0.0693 × expression of LZTS2) + (0.2521 × expression of SCUBE3) + (0.1988 × expression of MAPK8IP2) − (0.5015 × expression of FOXO6). The patients in the training cohort were divided into a high-risk group (n = 22) and a low-risk group (n = 22) according to the median cut-off value. The Kaplan–Meier curve indicated that patients in the high-risk group had significantly worse DMFS than their low-risk counterparts (Figure 5A). The predictive performance of this four-gene signature model for DMFS was evaluated by a time-dependent ROC curve, and the AUC reached 0.78 at 6 months and 0.75 at 1 year (Figure 6A).

The least absolute shrinkage and selection operator-based (LASSO-based) feature selection. (A) Tuning parameter (λ) selection in the LASSO model used 10-fold cross-validation via minimum criteria. (B) LASSO coefficient profile plot produced against the log (λ) sequence.
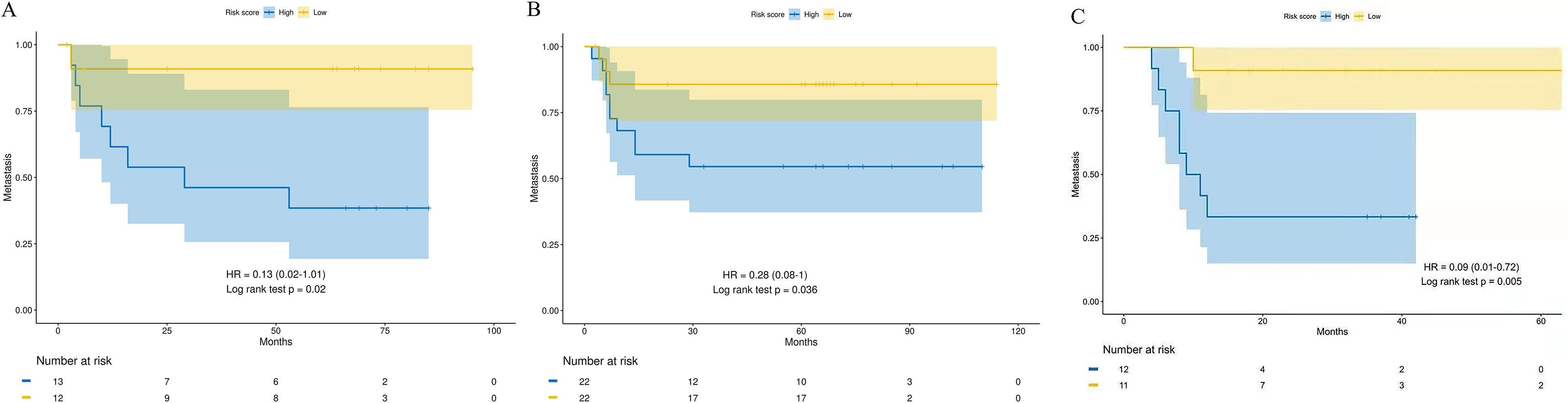
Kaplan–Meier curves of distant metastasis-free survival (DFMS) according to the four-gene signature model. (A) Nanning training cohort (n = 44), (B) Beijing validation cohort (n = 25), And (C) Wuzhou validation cohort (n = 23).
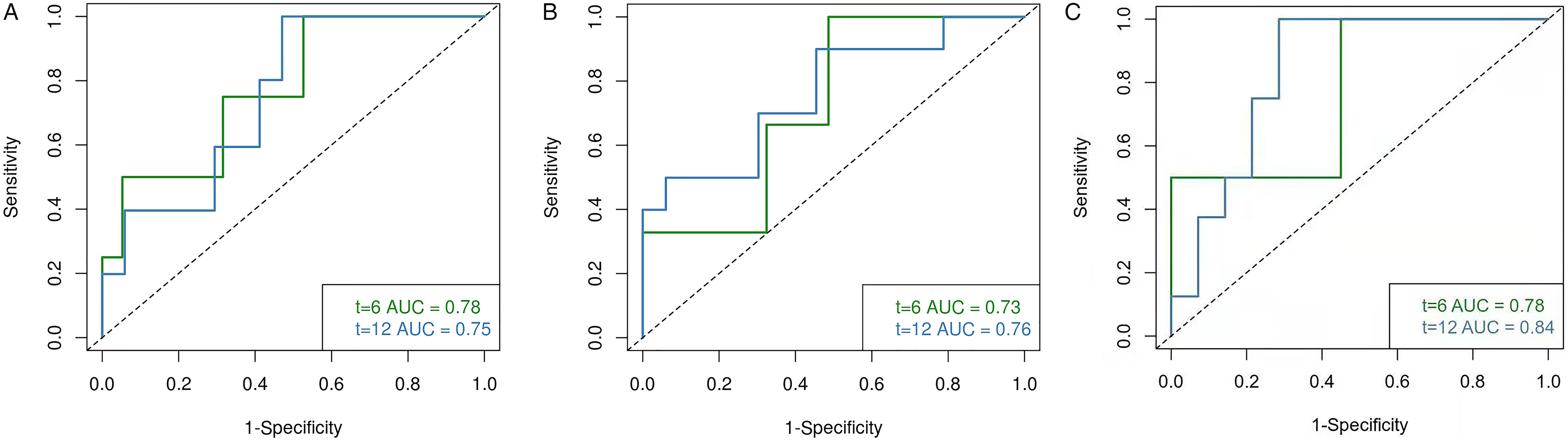
Time-dependent ROC curves of four-gene signature model for predicting the risk of distant metastasis. (A) Nanning training cohort, (B) Beijing validation cohort, And (C) Wuzhou validation cohort.
Validation of the Four-Gene Signature Model in the Validation Cohort
To test the robustness of the four-gene signature model constructed from the training cohort, the patients in 2 validation cohorts were also stratified into high- and low-risk groups by the median cut-off value calculated with the same formula derived from the training cohort. Likewise, the patients in the high-risk group were more likely to suffer from distant metastasis and had a shorter survival time than those in the low-risk group (Figure 5B, and C). Moreover, the AUC of the four-gene signature model in Beijing validation cohort was 0.76 at 6 months and 0.73 at 1 year (Figure 6B), and was 0.78 at 6 months and 0.84 at 1 year when it in Wuzhou validation cohort (Figure 6C). To verify the independence of prognostic effect of gene model and other clinical features, we integrated the samples of 2 validation cohorts and performed univariate and multivariate analysis using Cox proportional hazard model. As summarized in Table 2 and Table 3, the four-gene signature model was an independent predictor of DMFS.
Univariate Analysis with Cox Proportional Hazard Model for DMFS of NPC Patients.

Abbreviations: DMFS, distant metastasis-free survival; NPC, nasopharyngeal carcinoma.
Multivariate Analysis with Cox Proportional Hazard Model for DMFS of NPC Patients.
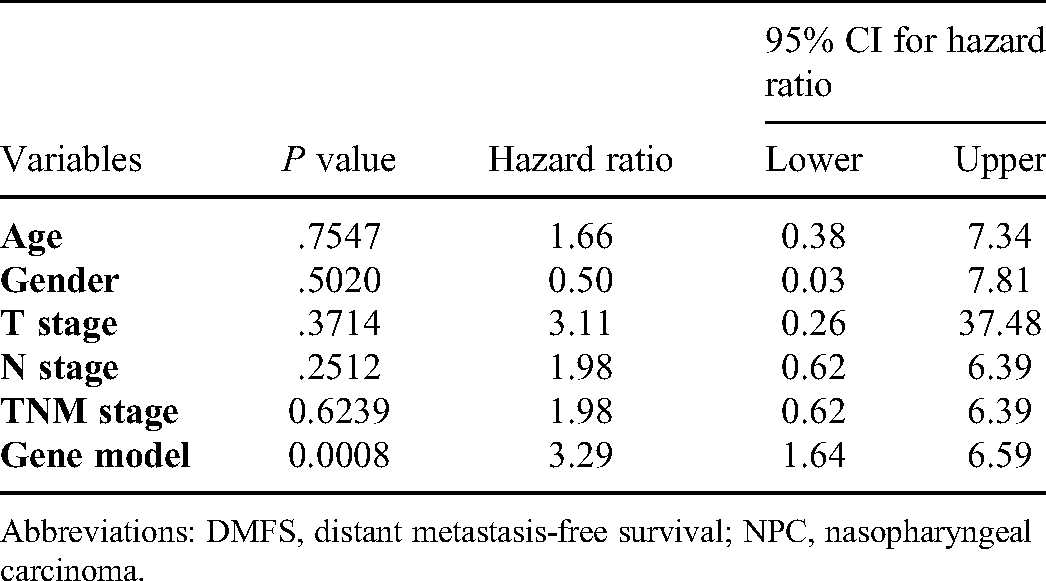
Abbreviations: DMFS, distant metastasis-free survival; NPC, nasopharyngeal carcinoma.
Discussion
High-throughput RNA-Seq is a sensitive and reliable technique that can detect aberrant genetic expression in cancer patients. 15 Although a few studies have explored and elucidated the DEGs or differentially methylated markers between noncancerous nasopharyngeal epithelium and NPC or between different NPC subtypes,16,17 the mRNA profiling differences between patients with different prognoses are still far from being understood. A recent study screened 137 DEGs between metastatic and nonmetastatic locoregionally advanced NPC samples and constructed a distant metastasis gene signature that consisted of 13 genes to predict the risk of distant metastasis in patients with locoregionally advanced NPC. 14 In the current study, a total of 1837 genes related to distant metastasis were identified, including 869 genes upregulated and 968 genes downregulated in patients with distant metastasis. Based on the identified DEGs, a novel prognostic model that can predict the risk of distant metastasis for patients with all stages of NPC was constructed in the Guangxi training cohort. A subsequent study showed that the novel prognostic model developed in the training cohort could categorize patients into high-risk and low-risk groups with significantly different metastasis-free survival rates. Furthermore, the AUC of this model was 0.78 and 0.75 at 6 months and 1 year of DMFS in the training cohort. Likewise, the AUC was 0.73 and 0.76 at 6 months and 1 year of DMFS in Beijing validation cohort, and was 0.78 at 6 months and 0.84 at 1 year when it in Wuzhou validation cohort. The multivariate analysis shows that the four-gene signature model was an independent predictor of DMFS.
The prognostic model proposed in this study was composed of 4 candidate genes (eg SCUBE3, LZTS2, MAPK8IP2, and FOXO6). Signal peptide-CUB-EGF-like domain-containing protein 3 (SCUBE3) is a secreted cell-surface glycoprotein involved in promoting epithelial–mesenchymal transition (EMT), angiogenesis, and metastasis in lung cancer by activating the transforming growth factor β (TGF-β) receptor-Smad2/3 pathway. 18 Overexpression of SCUBE3 results in the activation of TGF-β1, which can regulate the expression of TWIST1 and thus contribute to the EMT and distant metastasis of breast cancer cells. 19 To our knowledge, there is currently no related study focusing on the correlation of SCUBE3 and invasion or metastasis in NPC. Herein, our study suggests the possibility that the expression of SCUBE3 may be associated with the distant metastasis of NPC for the first time. MAPK8IP2 encodes a scaffold protein termed mitogen-activated protein kinase 8 interacting protein 2, which has been shown to interact with and regulate the activity of the MAPK8/JNK1 and MAP2K7/MKK7 kinases. In terms of cancer progression, the combined expression of MAPK8IP2 and its interacting proteins (FGF12 and MAPK13) has been found to correlate with poor survival in patients with esophageal squamous cell carcinoma. 20 FOXO6 is a member of the Forkhead transcription factor family (FOXO family) and encodes a transcription factor named FOXO6. FOXO6 has previously been identified to regulate memory consolidation and synaptic function. 21 Moreover, several studies found that FOXO6 was involved in the regulation of very low-density lipoprotein production by promoting gluconeogenesis and integrating insulin signaling with microsomal triglyceride transfer protein.22,23 However, the molecular function of FOXO6 in the oncogenesis and progression of various cancers remains controversial. Hu et al. 24 reported that overexpression of FOXO6 can inhibit the proliferation of lung cancer cells by inducing p53 accumulation, which indicates the tumor suppressor role of FOXO6. Conversely, Li et al. 25 found that FOXO6 was upregulated in gastric cancer and that overexpression of FOXO6 promoted cell proliferation by inducing C-myc expression. Therefore, the molecular roles of FOXO6 may be cell- or tissue-specific. The LZTS2 gene is located on human chromosome 10q24.3, and its gene product has previously been reported to inhibit the cell growth and proliferation of NPC cell lines in vitro and in vivo, which suggests that LZTS2 may function as a tumor suppressor in NPC.26,27 However, the effects of LZTS2 expression on the invasion and migration abilities of NPC cell lines were not elucidated in these studies. It is worth noting that according to the regression coefficient of LZTS2 in our present study, patients with higher expression levels of LZTS2 are more vulnerable to developing distant metastasis. In consideration of the contradictory outcomes above, we speculated that the role of LZTS2 may vary during different periods of NPC progression, and the same phenomenon has been confirmed in TGFβ. 28 During the early stages of various cancers, TGF-β has been reported to inhibit cancer cell proliferation by inducing the synthesis of CDKIs, p21 protein and 4EBP1 and suppressing the expression of the MYC gene.29–32 In regard to distant metastasis, the binding of TGF-β to its receptor TGFβ2R induces the phosphorylation of the PAR6 protein and the degradation of the RhoA protein, thereby disrupting the intercellular junction between cancer cells, accelerating the process of EMT and eventually promoting the development of distant metastasis.33,34
To gain more biological insight into the gene networks underlying distant metastasis in NPC, we performed GSEA and discovered that 6 gene sets associated with the Wnt/β-catenin signaling pathway, Hh signaling pathway, Notch signaling pathway, mitotic spindle, apical surface, and estrogen response late were enriched in NPC patients with distant metastasis. Of note, the Wnt/β-catenin signaling pathway has been reported to be involved in EMT and radioresistance in various cancers.35,36 A recent study indicated that the positive expression of 2 Wnt/β-catenin signaling pathway-related proteins (β-catenin and c-MYC) was negatively linked with the survival rate of NPC patients. 37 Moreover, activation of the Wnt/β-catenin signaling pathway leads to NPC cell radioresistance and metastasis through nuclear translocation of β-catenin and transcriptional upregulation of HR pathway-related and EMT-related gene expression. 38 The Hh signaling pathway was first identified in the fruit fly and exerts its biological effects through a signaling cascade that culminates in a change in the balance between the activator and repressor forms of glioma-associated oncogene (Gli) transcription factors. 39 According to recent reports, the Hh signaling pathway is involved in the development of at least one-third of all malignant tumors 40 and therefore provides new insight into the development of molecular targets and tumor prevention strategies associated with the Hh signaling pathway. For instance, when surgery and radiotherapy are not effective treatment modalities, targeted Hh signaling pathway inhibition has been regarded as the treatment for locally aggressive basal cell carcinoma and metastatic basal cell carcinoma. 41
Our study had some limitations. First, the sequencing data from 2 validation cohorts contained only 25 and 23 samples, respectively, and the current findings still need to be confirmed with a larger sample size. Second, the biological mechanisms by which the 4 candidate genes included in this prognostic model contribute to NPC metastasis remain unknown, and further assessment of their biological functions might provide novel targets for NPC treatment.
Conclusion
Our study identified 1837 DEGs and 6 gene sets that may be involved in the distant metastasis process of NPC, and we further developed a novel four-gene signature model that can assess the risk of distant metastasis in NPC patients. This model may be used as a new tool to predict the prognosis of NPC patients.
Footnotes
Abbreviations
Acknowledgments
The authors would like to thank Huimin Wen, Yu Luo, Diange Li, and Huanxi Li for performing the sample preparation and RNA-Seq experiment. We also thank Jinsheng Tao for performing the statistical analysis.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received funding through Guangxi Zhuang Autonomous Region Key Research and Development Program (Grant No. AB19259002).
Ethical Statement
The present study was approved by the ethical committee of People's Hospital of Guangxi Zhuang Autonomous Region (No: KY-2019-001), Xuanwu Hospital (CLE2021-153), and Wuzhou Red Cross Hospital (S2019-35). All patients provided written informed consent prior to enrollment in the study.