
Abstract
Background:
Previous studies reported that N-myc downstream-regulated gene 1 (NDRG1) was upregulated in various cancer tissues and decreased expression of miR-188-3p and miR-133b could suppress cell proliferation, metastasis, and invasion and induce apoptosis of cancer cells. However, the molecular mechanism of NRDG1 involved in hepatocellular carcinoma (HCC) tumorigenesis is still unknown.
Methods:
The expressions of miR-188-3p, miR-133b, and NRDG1 in HCC tissues and cells were quantified by qRT-PCR and Western blot. MTT assay and transwell invasion assay were performed to evaluate cell growth and cell migration, respectively. Luciferase reporter assay were performed to determine whether miR-188-3p and miR-133b could directly bind to NRDG1 in HCC cells.
Results:
The results showed that NRDG1 was upregulated and these 2 microRNAs were downregulated in HCC tissues. NRDG1 was negatively correlated with miR-188-3p and miR-133b in HCC tissues. MiR-188-3p and miR-133b were demonstrated to directly bind to 3′UTR of NRDG1 and inhibit its expression. Upregulation of miR-188-3p and miR-133b reduced NRDG1 expression in hepatocellular carcinoma cell lines, which consequently inhibited cell growth and cell migration.
Conclusions:
Our finding suggested that miR-188-3p and miR-133b exert a suppressive effect on hepatocellular carcinoma proliferation, invasion, and migration through downregulation of NDRG1.
Introduction
At present, hepatocellular carcinoma (HCC) remains one of the deadliest forms of cancer in the world, and yet no curative therapies exist for individuals with advanced forms of this disease. 1 HCC tumors exhibit high rates of genomic instability that can lead them to become highly aggressive and metastatic, resulting in their being the 4th most common cause of cancer-associated death globally. Despite the serious nature of this disease, the molecular mechanisms governing HCC development and progression are incompletely understood. In particular, the identification of the genes that serve as a molecular switch governing uncontrolled HCC cell growth is essential in order to guide efforts to disrupt HCC progression, and at present, the identity of such master regulators of HCC progression remains uncertain.
N-myc downstream-regulated gene 1 (NDRG1) is an NDRG family gene that has been shown to play key roles in cellular growth, differentiation, responses to stress, and hormone responses. 2 Previous reports have highlighted the overexpression of NDRG1 in gastric cancer, cervical cancer, renal cancer, and squamous cell carcinoma, suggesting it may play a role in driving the development or progression of these cancers. 3 -5 Indeed, altered NDRG1 expression levels have been detected in liver cancer, with the degree of NDRG1 expression being positively correlated with carcinogenesis and suggesting that NDRG1 targeting may be an ideal means of treating and curing these cancers. 6,7 For example, in past studies reductions in the expression of NDRG1 have been linked with increases in overall survival (OS), 8 -11 whereas increases in the expression of this gene have been found to correlate with the poorer OS. 4,6,12,13 Still, other researchers have highlighted the potential relevance of NDRG1 as a potentially valuable prognostic biomarker in cancer patients in addition to its role in the context of tumorigenesis. 3,14,15
MicroRNAs (miRNAs) are small RNA molecules (∼22 nucleotides) that lack coding potential and yet can regulate a wide variety of target genes at the post-transcriptional level via directly interacting with mRNA molecules and inhibiting their translation or even mediating their degradation. 10 Countless studies have documented the roles for specific miRNAs in regulating key processes including apoptosis, metabolic activities, cellular differentiation, and tumor invasion and metastasis. 16 Given their close association with specific pathways linked to oncogenesis, certain miRNAs may thus represent ideal biomarkers of carcinogenesis and/or viable therapeutic targets in cancer. Of particular note, both miR188-3p and miR-133b have been identified as putative tumor suppressor miRNAs in gastric, lung, and cervical cancer owing to their abilities to specifically target certain oncogenes. 17 -19 Many tumor suppressor miRNAs, which normally constrain proto-oncogene expression, are downregulated in cancer, and as such the introduction of synthetic miRNA mimics of such tumor suppressor miRNAs may be an ideal means of treating patients suffering from both solid and hematological cancers. The role of such tumor suppressor miRNAs in HCC, however, remains poorly studied and warrants further investigation.
In the present study, we therefore assessed NDRG1 expression in HCC and assessed the relationship between the expression of this gene and that of miR188-3p and miR-133b. We observed significant increases in NDRG1 expression in human HCC clinical samples, with the expression of this gene being inversely correlated with that of miR188-3p and miR-133b, which are known to suppress NDRG1 expression. When HCC cells were treated so as to increase miR188-3p and miR-133b expression, this led to a marked reduction in NDRG1 expression and substantial inhibition of the migratory and proliferative activity of these tumor cells. Together, these results suggest that miR-188-3p and miR-133b can regulate NDRG1 at the post-transcriptional level in HCC, indicating that the therapeutic use of mimics of these miRNAs may be a promising means of targeting NDRG1 and constraining tumor growth in affected patients.
Materials and Methods
Patient Sample Collection
In total, we collected 43 HCC patient tumor samples and matched paracancerous tissue samples (from the marginal zone >5 cm from the tumor) from the Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China). Samples were only collected from patients who had not undergone any specific tumor treatments prior to surgical biopsy or resection. A pathologist confirmed the diagnosis of HCC for all tumor samples. All experiments involved in human specimens were approved by The Research Ethics Committee of Wuhan Central Hospital (approval NO.19M031). All patients provided written informed consent prior to enrollment in this study.
Cell Culture
The HepG2 HCC cell line was from the Cell Bank of the Chinese Academy of Sciences and was grown in DMEM containing 10% FBS in a 5% CO2 incubator at 37oC.
Western Blotting
RIPA buffer supplemented with protease inhibitors, PMSF (1 mM), Na3VO4 (1 mM), and NaF (1 mM) was used for cell lysis, after which a BCA assay (Rockford, IL, USA) was used to assess protein levels in samples. Equal protein contents were then separated via SDS-PAGE and transferred onto a polyvinylidene fluoride (PVDF) membranes (#ISEQ00010, Millipore, MA, USA) that were then blocked in 5% skim milk prepared in TBST. Blots were then incubated overnight with primary antibodies specific for GAPDH (#5174, CST, USA) or NDRG1 (10893-1-AP, Proteintech, Wuhan, China) at 4oC. After washing in TBST, the membranes were visualized by enhanced chemiluminescence kit (#RPN2232, Amersham, GE Health, UK) and photographed.
qRT-PCR
Primers specific for genes of interest and GAPDH were synthesized by Sangon (Shanghai, China) Table 1, while appropriate sequence-specific primers corresponding to miRNAs of interest and the control U6 snRNA were also generated (Table 1). M-MLV (Promega, USA) or a miRNA reverse transcription kit (Qiagen, USA) were used based on provided directions in order to mediate RNA reverse transcription, after which qRT-PCR (BioRad, CFX96, USA) was conducted with the isolated cDNA. Thermocycler settings were as follows: 95oC for 10 min; 40 cycles of 95oC for 15 s, 65oC for 5 s, and 72oC for 45 s. Gene expression for mRNAs and miRNAs of interest was determined based upon the Ct values for each gene following normalization to GAPDH and U6 controls, respectively, with triplicate samples analyzed in all analyses.
Primers Used in This Study.

Transfection
Cells were plated at 50% confluence for 16 h, after which they were transfected using 20 ul Opti-MEM (Invitrogen, USA) containing appropriate miRNA or RNAi constructs, after which downstream assays were performed as indicated. miRNA mimics and miR-NC were all obtained from Sangon Biotech (Shanghai, China).
Luciferase Assays
The 3′-UTR of the NDRG1 mRNA was amplified and was then cloned into the pGL3 vector using the primers shown in Table 2, with Xba1 digestion and CIP treatment used to remove 5′ phosphates from the vector prior to ligation to prevent self-assembly. Sequencing was used to confirm the identity and orientation of the pGL3-NDRG1 3′UTR vector _ HepG2 cells were then co-transfected with this vector together with the control TK vector and appropriate miRNAs or other constructs as in past studies. 20 After 24 h, luciferase activity was assessed based on provided directions. Dual-Luciferase Assay System from Promega (Madison, WI, USA) was used to measure the activities of firefly luciferase and Renilla luciferase in the cell lysates.
miRNA Mimics and Antago-miRNA.

MTT Assay
Cell proliferation was analyzed via an MTT assay. Briefly, treated cells were added to 96-well plates (1000/well), after which 100 μg/well MTT (3-(4,5-methylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide) (Servicebio, Wuhan, China) was added per well for 4 h. After this time, DMSO was used to solubilize formazan products in cells, and absorbance at 490 nm was assessed. Samples were analyzed in triplicate.
Cell Migration Assay
Cells were first collected and were then resuspended in serum-free media, after which they were added to the upper chamber of a transwell insert (BD, Biosciences, San Jose, CA, USA, 8 µm pore size; 5 × 104 cells/chamber). Media that contained 10% FBS was added to the lower chamber as a chemoattractant for these cells. Cells were then incubated for 6 h, after which 3.7% formaldehyde was used to achieve cell fixation followed by staining of cells with crystal violet. Non-migratory cells were then carefully removed using a cotton swab, whereas cells that had migrated into the lower chamber were quantified via microscopic analysis.
Data Analysis
Prism 6.0 (Graphpad Software, CA, USA) was used for statistical testing. Data are means ± SD and were compared via Student’s t-tests, 2-way ANOVAs, or repeated measures ANOVAs as appropriate, with P < 0.05 as the significance threshold.
Results
The Demographic, Clinical Characteristics of the Study Population
The age of the 43 patients ranged from 43 to 76 years with a mean of 56.3 (±11.3) years. The ratio of men to women was 4.38 (35/8), and 26 of 43 patients had a family history of HCC, 31 patients had chronic HBV infection, 1 patient was positive for hepatitis C virus RNA, and 19 patients had cirrhosis of the liver. The median tumor size was 45 mm (IQR 25-68). The number of diagnosed cases of AJCC stage T1, T2, T3, and T4 disease was 25, 7, 6, and 5, respectively.
HCC Patient Samples Exhibit Increased NDRG1 mRNA and Protein Levels
While previous studies have highlighted increased NDRG1 expression in HCC and other cancer types, 14,21 further studies of its baseline expression in HCC are still needed. To that end, we obtained HCC tumor tissue and paracancerous normal tissue samples and analyzed NDRG1 expression therein. We found that there was relatively limited NDRG1 expression in healthy normal tissue, whereas its expression was markedly enhanced in cancerous tissue samples (Figure 1A). To further explore the basis for this increased NDRG1 expression, we analyzed NDRG1 mRNA expression in these samples, again revealing increased expression of this gene in cancerous tissues relative to normal control samples (Figure 1B). The magnitude of this difference, however, was less pronounced than at the protein level suggesting that post-transcriptional regulatory mechanisms may further lead to enhanced protein level increases in NDRG1 expression in HCC.
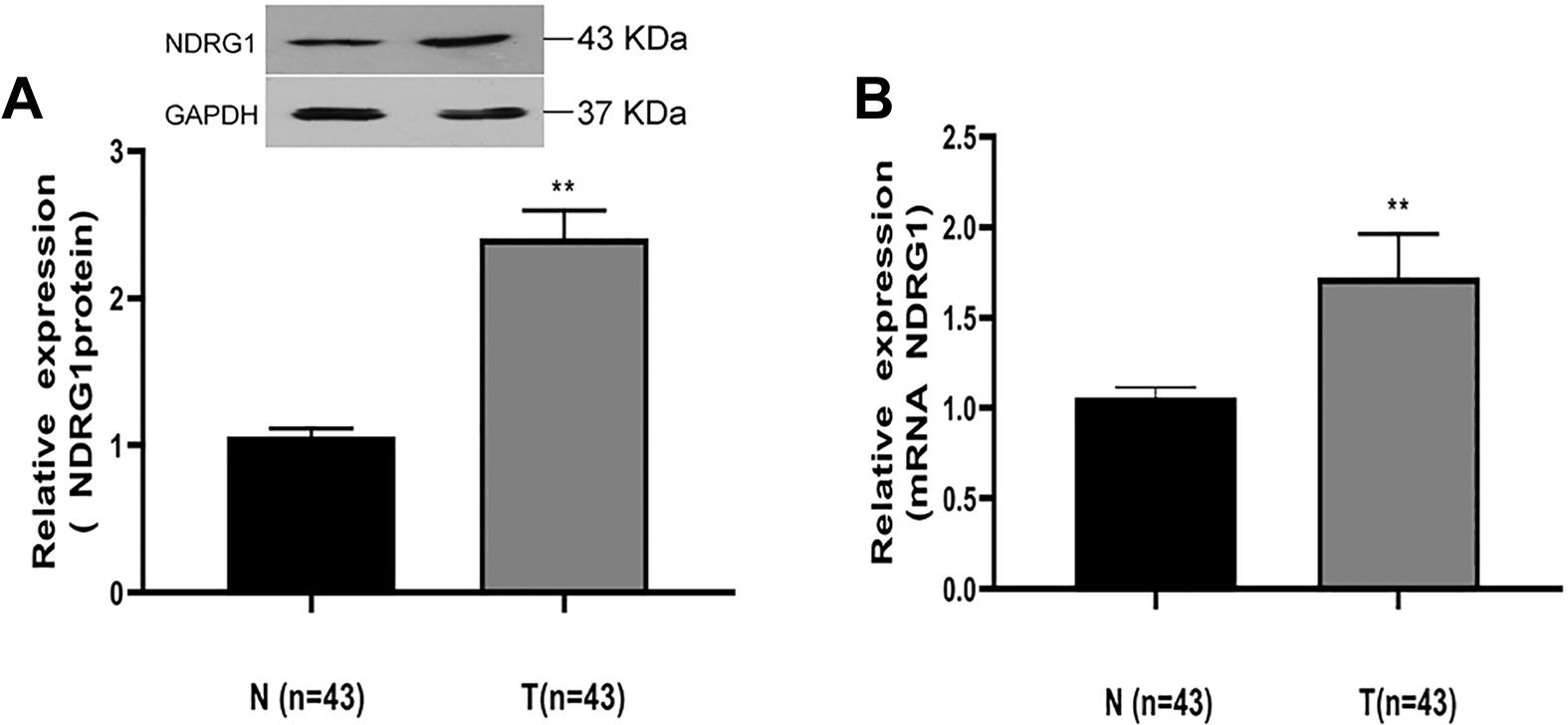
HCC patient samples exhibit increased NDRG1 expression. A, Western blotting was used to assess NDRG1 protein levels in 43 paired patient HCC samples and paracancerous control samples, with GAPDH as a loading control. B, Expression of NDRG1 at the mRNA level was assessed in the 43 indicated patient samples via qRT-PCR, with GAPDH as a normalization control. Expression was normalized to that in normal tissues. Data are means ± SD (** P < 0.01).
miR188-3p and miR-133b Expression Is Negatively Correlated With That of NDRG1
Given this possibility of post-transcriptional NDRG1 regulation, we next sought to identify potential miRNA regulators of the NDRG1 3′-URT using the miRbase platform, which highlighted miR188-3p and miR-133b as potential regulators of this mRNA (Figure 2A). To confirm this possibility, we therefore next assessed miR188-3p and miR-133b expression in the abovementioned tumor and adjacent control samples. We observed striking reductions in the expression levels of both of these miRNAs in cancerous tissue samples relative to paracancerous controls, with a clear negative correlation between their expression and that of NDRG1 in tumor tissues (Figure 2B and C). We therefore next examined whether miR188-3p and miR-133b were able to directly target the NDRG1 mRNA in HCC as predicted via screening available HCC cell lines for expression of these miRNAs and NDRG1. Of the tested cells, we found the HepG2 line to exhibit moderate expression levels of all of these genes (data not shown), making this cell line ideal for genetic manipulation to explore the interactions between NDRG1 and miR188-3p/miR-133b. We therefore transfected HepG2 cells with either mimics or antagonists of miR188-3p and miR-133b and then assessed NDRG1 in transfected cells, confirming transfection efficiency via qRT-PCR (Figure 3A and B). Consistent with the bioinformatic predictions, we found that transfecting these HCC cells with mimics of miR188-3p or miR-133b resulted in a clear reduction in the expression of NDRG1 (Figure 2D and E). In contrast, when endogenous levels of miR188-3p or miR-133b were repressed using antagonistic constructs, NDRG1 expression levels rose in treated cells (Figure 2D and E). A luciferase reporter assay further confirmed that miR188-3p and miR-133b were both able to bind to the 3′UTR of NDRG1 and thereby repress its transcription, whereas reducing levels of these miRNAs resulted in increased NDRG1 reporter activity (Figure 2F).

NDRG1 expression is negatively correlated with that of miR188-3p and miR-133b. A, The Targetscan tool was used to assess the 3′-UTR of the NDRG1 mRNA to identify potential sites for miR188-3p and miR-133b binding. B, Expression of miR188-3p was assessed in patient samples from Figure 1 via qRT-PCR, normalizing expression to that of the U6 snRNA (** P < 0.01). C, Expression of miR-133b was assessed in patient samples from Figure 1 via qRT-PCR, normalizing expression to that of the U6 snRNA (** P < 0.01). D, HepG2 cells were transfected using a miR-188-3p mimic, antagonist, or appropriate control constructs, and then NDRG1 expression was assessed via Western blotting. E, HepG2 cells were transfected using a miR-133b mimic, antagonist, or appropriate control constructs, and then NDRG1 expression was assessed via Western blotting. F, HepG2 were co-transfected using a control TK-Renilla reporter, an NDRG1 3′-UTR luciferase reporter, and appropriate miRNA mimics or antagonists. After 24 h, luciferase activity was measured and normalized to Renilla activity (* P < 0.05). Data are means ± SD of triplicate analyses.

NDRG1 knockdown impairs cellular migration. A, Transfection efficiency of miR188-3p and its antagonist. B, Transfection efficiency of miR-133b and its antagonist. C, NDRG1 knockdown efficiency at 48 h post-transfection of HepG2 cells with appropriate RNAi or control constructs in a 6-well plate, as assessed via Western blotting. D, After transfecting HepG2 cells with appropriate siRNA or control constructs for 24 h (a, no treatment; b, siNC; c, si-NDRG1), transwell migration assays were performed revealing a significant impairment in cellular migration upon NDRG1 knockdown. Assays were repeated in triplicate. E, Statistical analysis of results from (B) (* P < 0.05).
miR188-3p and miR-133b Mediate the NDRG1-Dependent Inhibition of HCC Growth and Migration
Given that our results thus far suggested the possibility that miR188-3p and miR-133b may be linked with NDRG1 dysregulation in HCC, we next explore the direct functional impact of this regulatory relationship in HepG2 cells. RNAi-mediated NDRG1 knockdown led to reduced expression of NDRG1 in these HepG2 cells as expected (Figure 3C), and this in turn markedly reduced the migratory activity of these cells (Figure 3D and E). Similarly, transfecting these cells with a miR188-3p mimic impaired both the growth (Figure 4A) and migratory activity of these cells (Figure 4B and C). When miR188-3p was instead antagonized, the opposite impact on cellular growth and migration was observed. In contrast, when miR188-3p activity was antagonized in cells in which NDRG1 had first been knocked down, no significant changes in cellular proliferation or migration were observed (Figure 4A-C). This thus suggested that NDRG1 is a primary target of miR188-3p, mediating its tumor suppressor-like role in HCC. Consistent with this, comparable findings were observed upon knockdown of NDRG1 and miR-133b in HepG2 cells, suggesting that this miRNA is similarly important for HCC cell proliferation and migration (Figure 4D-F).

miR188-3p and miR-133b-mediated inhibition of HCC proliferation and migration is NDRG1-dependent. A, At 12 h post-transfection with the indicated combinations of NDRG1 siRNA and miR188-3p mimics or antagonists, HepG2 cells were plated in 96-well plates and assessed via MTT assay at the indicated time points, revealing that miR-188-3p impaired cell growth whereas antagonizing it had the opposite effect, while antagonism of this miRNA had no impact on proliferation when NDRG1 was also knocked down. B, Cells treated as in (A) were used for transwell migration analyses. C, Statistical analysis of data from (B). D, After being transfected with the indicated combinations of NDRG1 siRNA and miR-133b mimics or antagonists, HepG2 cells were analyzed via MTT assays at the indicated time points. E, Cells treated as in (D) were used for transwell migration analysis. F, Statistical analysis of data from (E). Data are means ± SD of triplicate analyses (* P < 0.05).
Discussion
At present, HCC remains the 4th deadliest cancer globally, 1 with conventional treatment strategies including hepatic resection and transplantation being unsatisfactory for many patients with more advanced disease. As such, a better understanding of the molecular mechanisms governing HCC cell growth and metastasis are vital in order to improve clinical understanding and treatment options for HCC patients. In this study, we provided novel evidence shown that miR188-3p and miR-133b can suppress NDRG1 expression in HCC, leading to suppression of cellular growth and migration. When these miRNAs became dysregulated, this resulted in enhanced NDRG1 expression and activity. Dysregulated NDRG1 activity has been detected in many cancer types, and the therapeutic targeting of NDRG1 has thus been highlighted as a potentially ideal strategy for treating patients with HCC and other forms of malignancies. 14,21 While these early results are promising, additional work is needed to understand the specific mechanisms governing NDRG1 upregulation in HCC in order to highlight potential strategies for therapeutic inhibition of this gene.
Recent work has clearly shown that miRNAs are often dysregulated in cancers and that this dysregulation can have a profound impact on oncogenesis in HCC and other tumor types. 22 The specific mechanisms governing oncogenesis in particular contexts, however, remains to be fully clarified. In this report, we demonstrated that both miR188-3p and miR-133b are downregulated in HCC relative to normal tissue controls. When overexpressed, both of these miRNAs were able to inhibit HCC tumor cell growth and migratory activity and to reduce NDRG1 expression, suggesting that both miR188-3p and miR-133b exhibit tumor-suppressive activities in HCC.
In line with our findings, previous studies have observed downregulation of both miR188-3p and miR-133b in many cancer types consistent with their tumor-suppressive roles. Indeed, our results suggest that miR188-3p and miR-133b suppress tumor growth at least in part via targeting NDRG1, potentially in parallel as individual antagonism of either of these miRNAs alone failed to recapitulate the phenotype observed upon NDRG1 knockdown. It is important to consider that miRNAs are able to regulate myriad distinct target mRNAs, and as such both miR188-3p and miR-133b likely regulate many other target genes beyond NDRG1. While our results thus show that miR188-3p and miR-133b regulation of NDRG1 is a key mechanism governing HCC cell growth and proliferation, we cannot exclude the possibility that other targets of these miRNAs contribute to our observed phenotypes.
However, there still existed several limitations in our study. The sample size of our study is small and the relationship between these miRNAs needs to be further explored, the inhibitory effect of miR-188-3p transfection alone on NDRG1 was not significantly different compared with miR-133b transfection alone. Our current study has not clarified whether miR-188-3p and miR-133b can both act to inhibit NDRG1 expression, and which of them is more dominant and critical needs to be further elucidated in our future studies, such as cotransfecting them to observe their effects on NDRG1 expression.
We assume that these miRNAs have other target mRNAs which are tumor promoters and overexpression of these miRNAs hinders the expression of these mRNAs driving to a much better antitumor effect. Be that as it may, its potential component remains to be assist explored, and affirmed. Since numerous previous studies report the involvement of NDRG1 in ERK/MMP-9 pathway, 23 further investigation is warranted to study the pathways regulated by NDRG1 in HCC and to delineate its relationship with miR-188-3p and miR-133b.
Post-transcriptional regulation often serves as a negative or positive feedback mechanism in the context of cancer growth, 24 and as such the targeting of the mechanisms underlying this feedback circuit is vital to prevent tumor propagation. As they are very small, miRNAs may represent ideal therapeutic mechanisms for disrupting such regulatory circuitry given that they can be delivered to cells more easily than can larger proteins or nucleic acid constructs, mediating their regulatory activity immediately upon cellular entry.
Supplemental Material
Supplemental Material, sj-pdf-1-tct-10.1177_15330338211033074 - MiR-188-3p and miR-133b Suppress Cell Proliferation in Human Hepatocellular Carcinoma via Post-Transcriptional Suppression of NDRG1
Supplemental Material, sj-pdf-1-tct-10.1177_15330338211033074 for MiR-188-3p and miR-133b Suppress Cell Proliferation in Human Hepatocellular Carcinoma via Post-Transcriptional Suppression of NDRG1 by Zhenzhao Luo, Yue Fan, Xianchang Liu, Shuiyi Liu, Xiaoyu Kong, Zhonghuan Ding, Ying Li and Liqing Wei in Technology in Cancer Research & Treatment
Footnotes
Authors’ Note
Zhenzhao Luo, Yue Fan, and Xianchang Liu contributed equally to this work. The data are available upon reasonable request. All experiments involved in human specimens were approved by The Research Ethics Committee of Wuhan Central Hospital (approval no. 19M031). All patients provided written informed consent prior to enrollment in this study.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Health Commission of Hubei Province scientific research project (WJ2019M031) and Excellent Youth Foundation of Health and Family Planning Commission of Wuhan Municipality (WX17Q10).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.