
Abstract
The clinical use of molecular tumor profiling (MTP) is expanding and there is an increasing use of MTP data to manage patient care. At the University of Colorado, 18 patients were diagnosed with primary serous ovarian cancer between 9/2015 and 6/2019 and consented for banking and analysis of tumor, ascites and plasma. All 18 patients had tumor and plasma samples that were sent for MTP, and 13 of 18 patients additionally had ascites collected and sent for MTP. 50-gene panel testing and BRCA testing were performed on primary tumor. BRCA genetic variants were more likely to be identified in plasma as compared to ascites or tumor, though not statistically significant (P = 0.17). Co-occurring genetic variants between plasma and ascites were less common in comparison to co-occurring variants between tumor and plasma or tumor and ascites, though not statistically significant (P = 0.68). Variants in KDR (VEGFR2) and TP53 were most likely to be conserved across all 3 biocompartments. Mutant allele frequencies (MAF) of individual genetic variants varied across biocompartments, though tended to be highest in the tumor, followed by ascites.
Background
Ovarian cancer is the most lethal gynecologic malignancy, with over 295,400 new diagnoses and 184,000 deaths per year, globally. 1,2 While most patients with high grade serous ovarian cancer (HGSOC) will respond to first-line treatment—cytoreductive surgery and platinum/taxane based chemotherapy—over 80% will recur and develop therapy-resistant disease. 3 The 5-year overall survival rate of patients with advanced stage HGSOC is near 30%. 4 With the advent of readily available high throughput sequencing, molecular tumor profiling (MTP) has lent the possibility of classifying tumors based on genomic alterations rather than histopathologic characteristics. Understanding this disease further on a molecular level is paramount to broaden the application of precision medicine and ultimately extend the patient progression-free and overall survival.
American Society of Clinical Oncology (ASCO) recommends germline BRCA (gBRCA) and, if negative, somatic BRCA testing, at the time of confirmed diagnosis of epithelial ovarian cancer, regardless of family history. 5 Germline and somatic testing not only inform disease prognosis but guide treatment with poly (ADP) ribose polymerase inhibitors (PARPi) as maintenance therapy, contributing to an increase in progression-free and overall survival. 6 -10 While PARPi were initially approved in patients with gBRCA variants, 7 they are now approved in patients with an underlying homologous recombination deficiency (HRD), 10 including somatic BRCA mutations, and most recently in patients with a partial or complete response to platinum-based primary chemotherapy. 8
Based on The Cancer Genome Atlas, up to 50% of ovarian cancers have HRD 11 with 13.2% to 15.3% attributed to BRCA1/2 variants. 12,13 Despite the clinical impact of genetic testing in the ovarian cancer patient, only one third of patients with ovarian cancer complete genetic testing. 14
Beyond HRD and BRCA variants, molecular tumor profiling can guide other treatment options in the recurrent setting based on molecular aberrations. Despite guidelines for molecular tumor profiling outside of BRCA or HRD being less concrete, molecular tumor profiling continues to be an important aspect of understanding and treating ovarian cancer. However, the molecular heterogeneity within and between tumors in a single patient with ovarian cancer poses a challenge to the treatment and understanding of this disease. 15 As clonal populations diverge within a tumor, it is unlikely a single sample from primary tumor is representative of the total mutational burden. 16 Similarly, sequencing of cell free DNA (cfDNA) from different biocompartments, such as ascites and plasma, is less understood. 17 Assessing mutation allele frequency (MAF), the percentage of a mutant allele across all reads from that genomic region, is an approach to better define tumor heterogeneity and to provide a more comprehensive understanding of the disease.
In the present study, we aim to identify the genetic variants present in tumor, ascites and plasma from a single point in a patient disease course. Secondarily, we propose to evaluate the presence of conserved variants and mutation allele frequency in these 3 biocompartments.
Methods
Regulatory Compliance and Study Patients
With Institutional Review Board approval from the University of Colorado (COMIRB numbers 07-935/18-0119), 18 patients with serous ovarian cancer diagnosed between 9/2015 and 6/2019 were included in the study. Patients were required to have 1 year of follow up after diagnosis and primary surgery to be eligible. Clinicopathologic data was extracted from the electronic medical record. Tumor stage was determined by the International Federation of Gynecology and Obstetrics (FIGO) system.
Sample Collection and DNA Sequencing
Specimens from each patient biocompartment (tumor, ascites, plasma) were collected and prepared by the University of Colorado Gynecologic Tumor and Fluid Bank. Molecular tumor profiling and analysis was donated and completed by Circulogene (https://circulogene.com). DNA extraction and next generation sequencing (NGS) are previously described. 18 Circulating cell free DNA (cfDNA) was isolated using Circulogene’s proprietary Linear In Situ Amplification (LISA) cfDNA enrichment and recovery technology. Quantification was performed using the Qubit 2.0 Fluorometer with dsDNA BR and HS assay kits (Life Technologies). Ultra-deep targeted sequencing of cfDNA, tumor DNA or ascites DNA was performed using the Ion Torrent NGS. The targeted sequencing libraries were generated using the Ion AmpliSeq Library kit 2.0 and Cancer Hotspot Panel v2 according to the manufacturer’s instructions (Life Technologies). The starting material consisted of 1-10 ng of cfDNA, tumor DNA or ascites DNA and each sample was analyzed using a CLIA-certified, CAP proficiency-supported clinical test. A selected 50-gene panel plus BRCA1 and BRCA2 genes of over hotspot somatic 3,000 variants was used [ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1, CTNNB, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAQ, GNAS, HNF1A, HRAS, IDH1, IDH2, JAK2, JAK3, KDR, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, VHL]. After amplification, primers were partially digested by the Fupa enzyme (Lifetech), then ligated to Lifetech ION XPRESS barcodes and purified using Ampure Beads. The quality of the libraries was assessed using quantitative PCR. The Ion Chef system was used for emulsion PCR to clonally amplify sequencing templates. NGS was performed on Ion Torrent Proton with coverage ranges of 3000-8000X. Two tiers of software validation (built-in VariantCaller 4.2 and built-out Station-X) were used for validation. Sequencing data was analyzed by the VariantCaller 4.2 software using the somatic high stringency parameters and the targeted and hotspot pipelines. All the variants identified were further confirmed by analyzing the data through GenePool (Station-X). Filter criteria was based on somatic and germline databases: COSMIC, dbGAP, 1000 Genomes, EXaC, and GNOMaD. Libraries were also generated from blank and control samples to ensure proper construction. Two cell line controls (SW480 and NA19240) and a “process control” with a true negative (from normal individuals who tested negative previously) were used to rule out false positives. The quality score threshold for reporting a variant was a minimum of 10. Any mutation allele frequency under 1% (below NGS platform detection limit) was filtered out.
Statistical Analysis
GraphPad Prism (v8.0) was utilized for descriptive statistics calculations and univariate analyses. Ordinary one-way ANOVA was utilized with a companion Brown-Forsythe test to study differences between the 3 biocompartments. A P value of less than 0.05 was considered significant.
Results
Demographics and Disease Characteristics
Eighteen patients were diagnosed with primary serous ovarian cancer between 9/2015 and 6/2019 and consented to tissue and fluid banking by the University of Colorado Gynecologic Tumor and Fluid Bank. Median age at diagnosis was 61 (range 48-80) at the time of diagnosis. The majority of patients were White (n = 16, 89%) and the remainder identified as Asian (n = 2, 11%). The majority of patients were diagnosed with at least stage IIIC disease (n = 11, 61%). Ninety-four percent of tumors were high grade (n = 17).
Ninety-four percent of specimens were collected at time of primary diagnosis. Forty-four percent received neo-adjuvant chemotherapy (NACT), while 56% did not. Eight patients recurred during the study period; 6 of the 8 patients who recurred had received NACT. There were no deaths reported in the patient electronic medical record as of the time of final data analysis. Two patients were lost to follow up as 1 patient moved to another practice and a second patient did not return for oncologic follow up as scheduled but is alive at the time of data analysis as she presented for care unrelated to her cancer diagnosis (Table 1).
Patient Demographics and Disease Characteristics.

BRCA Testing
BRCA variants were more likely to be identified in plasma as compared to ascites or tumor, though not statistically significant (Ordinary one-way ANOVA, P = 0.17). Seven of the 18 plasma samples had BRCA variants identified. Three of the 7 had more than one BRCA variant identified; BRCA2 variants were more common than BRCA1. Five of the 18 tumor samples had BRCA variants identified. Only 2 of the 13 ascites samples sent for BRCA testing had variants identified. Two patients had a BRCA variant (BRCA2 c.5467A > T p.K1823*) identified in both tumor and plasma. There was no BRCA variant identified and conserved across all 3 biocompartments (Table 2).
BRCA Somatic Testing Mutation Identification.a

a Unknown = Not available, 0 = Negative, 1 = BRCA1, 2 = BRCA2; QNS, quantity not sufficient.
50-Gene Testing Panel
Ascites carried more genetic variants on average in comparison to tumor or plasma, though not statistically significant (3.62 v 2.44 v 2.28, Ordinary one-way ANOVA, P = 0.09). Point mutation in KDR Q472 H and TP53 P72 R were most likely to be conserved across all 3 biocompartments (Table 3). Other conserved genetic variants included the following: JAK3 V722I, MET T1010I, KIT M541 L and PIK3CA I391 M. The average number of co-occurring genetic variants between plasma and ascites was less in comparison to co-occurring genetic variants between tumor and plasma or tumor and ascites, though not statistically significant (1.2 v 1.4 v 1.5, Ordinary one-way ANOVA, P = 0.68).
Identifying Co-Occurring Mutations Using Somatic Testing 50 Gene Panel.

Mutation Allele Frequency
Mutant allele frequencies (MAF) varied among the 3 biocompartments (Table 4). The average MAF was highest in tumor, followed by ascites and then plasma (Figure 1A). Highly conserved variants including KDR Q472 H and TP53 P72 R tended to have higher MAF. Mutational profiles of representative patients depict the heterogeneous and most often discordant MAFs between the tumor, ascites, or plasma (Figure 1B; Table 4).
Mutation Allele Frequency in Patients With Mutations Identified in All Biocompartments.


Mutation allele frequency highlights the degree of mutational heterogeneity within a patient’s different biocompartments. A, Mean mutant allele frequency and standard error of the mean detected in plasma (n = 19), tumor (n = 19), and ascites (n = 13). B, Mutant allele frequency of 5 sample patients within plasma, tumor, and ascites. Connecting lines between plasma, tumor, and ascites are specific mutations (colored matched) indicated on the right of the graph.
Discussion
Molecular tumor profiling allows for the classification of tumors based on genomic alterations rather than histopathologic characteristics. While grade and histology do have prognostic implication, research in both gynecologic and non-gynecologic cancers have found the prognostic utility and application of precision treatment based on molecular alterations. 19,20
Though molecular tumor profiling is utilized regularly in melanoma 21 and lung cancer, 22 its utility in the treatment of ovarian cancer continues to be explored as research expands beyond PARPi and defects in homologous recombination, including BRCA. Current changes in gynecologic oncology, in general, include transitioning to a molecularly based profiling of cancer to aid in delineating a comprehensive treatment plan. For example, recently ASCO recommended women diagnosed with clear cell, endometrioid or mucinous ovarian cancer be offered testing for microsatellite instability, rendering them potentially sensitive to immune-checkpoint blockade. 5 Further integration of molecular classification in ovarian cancer is warranted.
There are a number of next generation sequencing platforms to assess mutational profiling, with most platforms having similar sensitivity. The pathologic relevance and understanding of genetic variants beyond BRCA1/2 are limited to only a few other genes. For instance, BRCA1/2-variants significantly increase the risk breast and ovarian cancer, but not all variants convey the same magnitude of risk and variants of unknown significance (VUS) are often identified. In a retrospective analysis of genetic testing from 83,000 breast and ovarian cancer patients, 8%-15% of variants were clinically “actionable.” 14 Further, the prevalence of VUS matched that of pathogenic variants. The differential impact of VUS versus pathogenic variants is an active area of research. As such, the scientific and clinical communities claim to have a better understanding of BRCA1/2 variants, highlighting how little is understood of other genetic variants. LaDuca et al observed that in BRCA1/2-associated cancers only 33.1% of patients had defined pathogenic variants of BRCA1/2 and 67% had mutations in other cancer associated genes. 23 Beyond BRCA1/2, most mutational profiling panels have up to 48 other genes and the clinical implications of the other variants needs further research.
Our data demonstrates that specimens from different biocompartments obtained from the same patient at a single time in their disease course harbor different genetic variants. Our study suggests that a more comprehensive view of the molecular tumor burden is achievable through sampling of different biocompartments.
This study is unique in that we evaluated different biocompartments, revealing that few genetic variants are conserved between primary tumor, ascites and plasma (Figure 2). Among the 13 patients with all 3 biocompartment samples, only 5 patients had conserved variants. The most commonly conserved genetic variants were in KDR and TP53. KDR mutant phenotype is associated with increased expression of vascular endothelial growth factor (VEGF); in a study of melanoma with germline KDR variants, cells that harbored this mutation were more invasive, with increased sensitivity to VEGFR2 antibody treatment. 24 TP53 variants are hallmark to many solid tumors, including ovarian cancer, often resulting in a loss of tumor suppressor function. To date, there are no molecularly matched therapies for either of these variants; however future studies examining in vitro and in vivo use of anti-angiogenic chemotherapies in the setting of KDR mutant tumors is of interest.
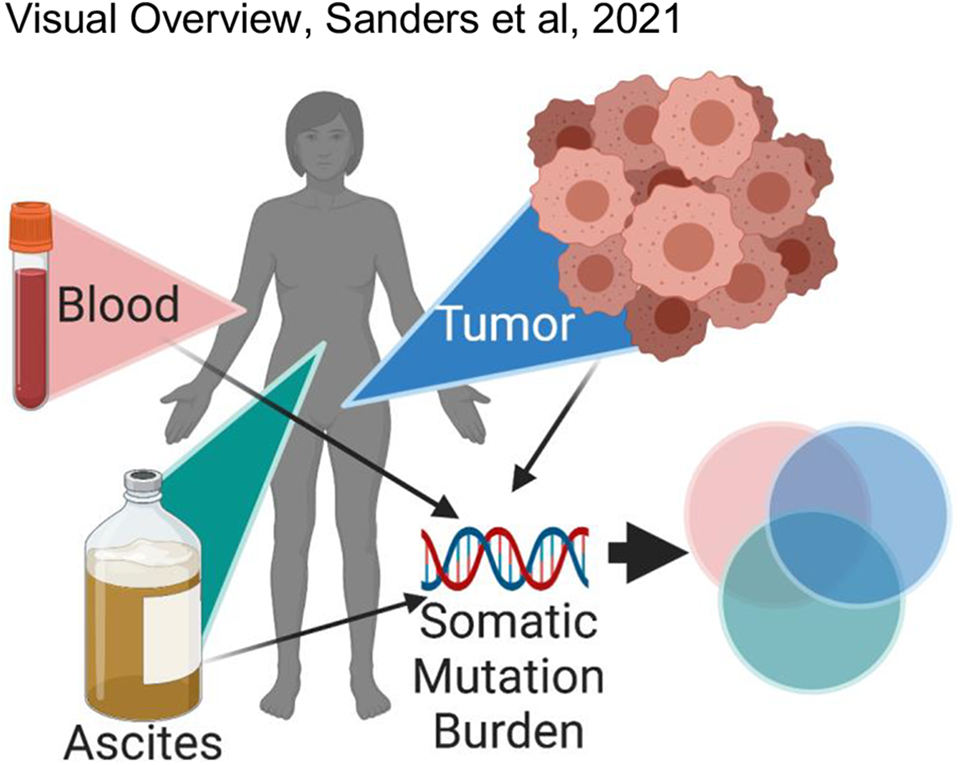
Visual Overview. Cross-sectional somatic genetic testing from diverse biocompartments within a matched patient. Cell free DNA isolated from blood and ascites fluid and DNA isolated from primary tumor used for next generation sequencing somatic BRCA testing and a 50-gene panel testing.
Additionally, this study found that BRCA variants were more commonly found in plasma in comparison to ascites or tumor, however, continuing this study on a larger scale is necessary to determine if this would be statistically significant. Should BRCA variants be more commonly identified in cfDNA of patient plasma, clinicians may start with an examination of plasma to determine BRCA variant status.
Mutant allele frequency (MAF) was also evaluated in this study. Interestingly, the average MAF was highest in tumor and ascites compared to plasma, perhaps signifying that despite heterogeneity within the various biocompartments, some variants may be required to maintain an oncogenic state. Completed on a larger sample size, understanding MAF of variants in different biocompartments may provide insight to the temporal sequence of mutations that facilitate disease progression. 25 While intriguing, the evolutionary significance underlying the biocompartment-specific genetic profiles is unclear. In spite of these gaps in knowledge, up to 15% of variants are clinically actionable. Thus as more molecular testing is completed, the signal to noise ratio involved in identifying novel pathologic variants will consequently improve.
Historically, clinical trials in oncology have focused on treatment for a particular tumor origin and histology. However, with the continued understanding of cancer on a molecular level, inclusion criteria will become more tumor agnostic. Beginning in rare tumors, the “basket” trial approach investigates a treatment in different primary tumors which share a common pathogenic variant. In comparison, “umbrella” trials evaluate different treatments based on molecular signatures, within a single tumor type. 26 While these designs acknowledge the molecular nuances of a tumor, our studies show that genetic variants are not always conserved in the different states of a cancer (ascites versus primary tumor). Sampling of various biocompartments, not just primary tumor, may lend itself to identify other mutational events. The development of “basket” and “umbrella” trials in the future may also include patients with either conserved variants or broaden its approach and include patients with a pathologic variant of interest, regardless of whether it was found in primary tumor, ascites or plasma.
Limitations to this study include a small sample size of only 18 patients with serous ovarian cancer with 44% of patients receiving NACT. Further, some of the variants identified may be germline in origin given the nearly 100% MAF for variants such as TP53 P72 R and KDR Q472 H. While TP53 P72 R and KDR Q472 H were thought to be benign polymorphisms, there is now data to support that TP53 P72 R results in a potentially worse prognosis for patients with ovarian cancer and KDR Q472 H is associated with a more aggressive phenotype in melanoma. 24,27 Additionally, this study did not specifically delineate variants as germline from somatic nor pathologic variants from VUS. Nonetheless, this study includes molecular profiling on 3 different biocompartments in patients with primary serous ovarian cancer. To our knowledge, there is a paucity of literature pertaining to the simultaneous examination of different biocompartments in this population, and our study provides additional data to support the further exploration of this approach.
Conclusion
In this single-institutional study of patients with primary serous ovarian cancer we evaluated the mutational burden across biocompartments using a BRCA and 50-gene panel. Conserved variants across all 3 compartments were rare. Our study indicates that the molecular signature of a tumor is complex and evolving. Expanded use of genomic testing should be encouraged and employed as precision medicine continues to progress, making treatment options and clinical trials available for our patients. Further studies should include collection of multiple specimens as patients continue through their treatment, disease free period and likely recurrence to better understand the dynamic molecular drivers of this disease. Larger studies powered for multi-variate analysis are also of interest.
Footnotes
Abbreviations
Authors’ Note
BS and BB were responsible for study design, data analysis and manuscript preparation. LK and PW provided critical revisions to the manuscript. All authors approved the final manuscript. The data are not available in a public database. The data generated are available upon request. This study was completed with the Institutional Review Board approval from the University of Colorado (COMIRB numbers 07-935/18-0119).
Acknowledgments
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr. Paul Walker is the Chair of Circulogene.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We acknowledge funding from The University of Colorado OB/GYN Division of Gynecologic Oncology (BGB). This work was supported by grants from the NIH/NCI (BGB, R00CA194318-03), Department of Defense Award (BGB, OC170228), the American Cancer Society Research Scholar Award (134106-RSG-19-129-01-DDC), and University of Colorado Cancer Center Support Grant (P30CA046934).