
Abstract
Solution Blow Spinning (SBS) has garnered significant attention for its rapid production of fibers from polymeric solutions. However, its efficacy is hindered by the low evaporation rates of aqueous solvents. In a groundbreaking development, we introduce the Heat Assisted Solution Blow Spinning (HA-SBS) system, which, for the first time, enables the one-step production of crosslinked Polyvinyl alcohol (PVA) fibers from an aqueous solution. By incorporating chlorhexidine (CHX) and/or graphene nanoplatelets (GNP) into the starting solution, we formulated four distinct variations. Our results reveal that all HA-SBS-produced systems maintain robust structural integrity and fibrous architecture after water immersion. The inclusion of GNP nanofillers not only enhances the crosslinking level but also significantly boosts mechanical performance. Release tests indicate that HA-SBS membranes effectively decelerate CHX release, providing controlled and sustained antimicrobial efficacy. Notably, CHX-containing membranes exhibit potent antimicrobial activity against both Gram-positive and Gram-negative bacteria. This innovative approach holds great promise for advancing the production of functional fibrous membranes, with broad implications for various applications.

Keywords
Introduction
Polymeric fibrous membranes are attracting considerable interest in many fields of application owing to their exceptional qualities, including high surface area, porosity, loading capacity, and flexibility.1–9 These membranes are produced by various process techniques, including phase separation, centrifugal spinning, wet spinning, self-assembly, direct drawing, melt blowing.10–15 However, the most frequently used technique is electrospinning (ES). Despite ES generate numerous advantages in terms of nanofibrous structure quality and control, it can be considered disadvantageous from an environmental and economic perspective. In fact, obtaining fibers via electrospinning requires long production times, energy consumption and poses potential risks for the operator. These drawbacks limit the industrial-scale production of nanofibrous membranes, resulting in a restricted application fields for such devices. Therefore, both academia and industries are focusing on developing more sustainable production methods that are environmentally and economically viable. To this end, among the possible existing alternatives to produce nanofibrous membrane, solution blow spinning (SBS) has demonstrated to be a processing technique comparable to electrospinning but with significantly reduced costs and production times.16–21 SBS involves the ejection of a polymeric solution through a system of concentric coaxial nozzles where the polymer solution flows in the core and compressed air flows in the shell. The mechanism forming fibers occurs due to the compressed air, which has a dual function of stretching/spinning the polymer solution and facilitating the evaporation of the solvent. Clearly, the air spinning mechanism enables an increase in production speed and a decrease in energy consumption compared to the electrospinning process, which is characterized by low er production times and high voltages.
Recently, Poly-vinyl-alcohol (PVA) fibers have attracted attention in biomedical, filtration, packaging22–33 applications, thanks to their distinctive characteristics, including biodegradability, water solubility, and excellent biocompatibility. Among the numerous potential applications, one that has generated significant interest is drug release.27,34–36 However, despite their excellent characteristics, PVA fibers, due to their highly hydrophilic behavior, immediately dissolve upon contact with aqueous media, which represents a disadvantage for sustained release. Generally, chemical or thermal treatments are carried out to overcome these limitations, in order to achieve good stability of the fibers and make them insoluble in aqueous media.24,37–43 However, chemical treatments are expensive in economic terms and they adversely affect the environment and human health because they require high energy expenditure and/or the use of cross-linking chemical agents.38,39,44
Only a limited numbers of works have been reported on the preparation of PVA membranes through the SBS technique. In fact, the slow evaporation rate of the PVA solvent (water) pose serious limit on the obtainment of fibers by SBS process. Moreover, based on the information available to us, PVA fibers were never produced and crosslinked in one step using the SBS technique.45,46 Medeiros et al. used a Bunsen burner to heat the air and force the solvent to evaporate at the nozzle outlet. Despite obtaining good fibrous structure, they were unable to cross-link the fibers. In other works, PVA was processed with SBS using solvents other than distilled water.17,47 Snari et al. succeeded in spinning a mixture of PVA and polylactide through solution blow spinning, but in this case, the addition of glutaraldehyde vapors was necessary to stabilize the membranes. 48
Considering the state of art, the idea of developing a process that allows the production and simultaneous cross-linking of PVA fibers could be truly interesting and could provide a significant advantage in terms of economic and environmental sustainability and/or biocompatibility. To this end, in this study an innovative SBS system, namely Heat Assisted Solution Blow Spinning (HA-SBS), it was implemented. Starting from a polymeric solution of distilled water (DI) and PVA, it was possible to achieve the production and simultaneous cross-linking of the fibers. The relationships between process-structure-properties of the obtained membranes were investigated, and a possible release application of Chlorhexidine (CHX), a rife hydrophilic antiseptic used in biomedical fields, was also evaluated. Based on the information available on scientific literature, such a setup had never been developed in other works.
Materials and method
Materials
Polyvinyl alcohol (PVA, Mw 89,000/98,000 Da, 99+% hydrolyzed) and chlorhexidine (CHX) diacetate salt C22H30Cl2N10 (C2H4O2) were procured from Sigma-Aldrich. Graphene nanoplatelets (GNP), were provided by XG Sciences Inc., Lansing, USA, MI. The structure of nanoparticle consists in a multiple layers of carbon nanotubes with thickness values approximately 10-20 nm, average width range 1 to 2 μm and average surface area of 750 m2/g. All the constituents were utilized as provided.
Production process of PVA fibers membranes
PVA (12 wt%) was dissolved in distilled water at temperature of 95°C with intense magnetic agitation for 4 h. For crafting the composite mats, CHX and/or GNP were incorporated into the polymer solution at 2 wt% and 1 wt%, respectively. (referred to polymer concentration). The PVA-based fibers membranes were obtained by heat-assisted Solution blow spinning technique (HASBS). The HASBS (Figure 1(a) and (b)) apparatus consisted in a compressed air source fitted with a pressure regulating device, a glass syringe, an electronic injection pump for regulating polymer solution flow, a spinning setup featuring concentric nozzles, a cylindrical plexiglass chamber, and a heated collector coated with aluminum foil. Figure 1 shows a schematic representation of HASBS. In detail, the coaxial needle with concentric nozzles is vertically placed near the plexiglass chamber. Coaxial needle is manufactured in AISI 316 stainless steel and consists of inner needle (inner diameter 0.45 mm, outer diameter 0.85 mm) concentrically placed in outer needle (i.d. 1.37 mm, o.d. 1.83 mm. The inner nozzle is injected with the PVA polymeric solution using a glass syringe installed on an electronic pump. Contextually, compressed air is blown into the outer nozzle. The blowing process takes place inside the plexiglass chamber, where fibers are formed and collected onto the hot collector. All solutions were processed with the same process parameters (shown in Figure 1(c)), in particular: flow rate of polymer solution = 45 mL/h, air pressure = 0.5 MPa, needle-to-hot plate collector distance = 20 cm, spinning operation = 15 min, temperature of plate collector = 180°C, temperature of chamber = 60°C, temperature at the nozzle tip = 60°C. Table 1 shows the sample codes and their formulations for all the systems used in this work. (a) Photo of the Heat assisted solution blow spinning (HA-SBS) device (b) Schematic image of HASBS (c) Processing condition. Specimens code and compositions of the corresponding solutions.

Morphological characterization
The morphological characterization of the fibers membranes was evaluated by Scanning Electron Microscopy, (SEM, Phenom ProX, Phenom-World), with an optical magnification range of 20–135x, maximal digital zoom of 12x, electron magnification range of 80–130,000x, acceleration voltages of 10 kV equipped with EDX probe to detect the presence of Cl signal. To analyze the membrane morphology, a Phenom Charge Reduction Sample Holder was used. This holder allows the examination of non-conductive samples without applying a conductive coating, providing high-quality images without compromising the integrity or altering the chemical/physical surface of the sample. The microscope is fitted with a sample holder under controlled temperature conditions (25°C). The specimens were obtained by cutting them off directly from the membranes prepared. The size distribution of fiber diameters was analyzed by Image J software, incorporating the Diameter J plugin. 36 For further information, refer to our earlier publication.37,38
Rheological characterization
The rheological tests of different PVA solutions were performed using a rotational rheometer (ARES/G2). A 25-mm parallel-plate geometry was used, and all tests were conducted under the following operating conditions: temperature of 25°C, frequency sweep mode in the range of 1–100 rad/s, with a constant stress of 1 Pa imposed.
Chemical analysis of surfaces
The chemical structure of fibers surfaces was examined by FT-IR/ATR analysis, using a Perkin-Elmer FT-IR/NIR Spectrum 400 spectrophotometer. The wavenumber range between 4000 and 400 cm−1 was considered for spectral analysis.
Mechanical characterization
The mechanical properties were explored through tensile tests (ASTM D882), conducted with a laboratory dynamometer provided with a 1 kN load cell (Instron 3365, UK). The assessments were carried out on samples (10 × 90 mm) cut off from the mats. The tests were performed using a dual crosshead speed: 1 mm/min for 2 min, followed by an increase to 50 mm/min until fracture occurred. The grip distance was 20 mm, whereas the sample thickness was measured before each measurement. Seven samples were examined for each material, the average values of the mechanical parameters and standard deviations were reported in particular, elastic modulus (E), tensile strength (TS), and elongation at break (EB).
Chlorhexidine kinetic release
A range of deionized water (DI) solutions, each containing a noted amount of CHX, was analyzed using a UV/visible spectrophotometer (model UVPC 2401, Shimadzu Italia s.r.l., Milan, Italy). This analysis aimed to establish a calibration curve, linking the intensity of the absorbance band of CHX with its concentration in DI water. The maximum absorbance band of CHX was identified at 230 nm. CHX release from the fibers was examined by submerging pre-weighed square samples. (10 × 10 mm2, approximately 0.01 g) in 10 mL of DI water at temperature of 37°C. The intensity of the absorbance bands in the solutions was measured at specific time intervals, and this data was used to determine the amount of CHX released by applying a calibration curve. Subsequently, after each measurement, all specimens were placed in 10 mL of clean DI water. Each measurement was carried out three times for accuracy.
Peppas-Korsmeyer model
The release data obtained were modelled using the Peppas-Korsmeyer equation:
Crosslinking level measurement
The crosslinking level of the samples was assessed by performing dissolution tests in deionized water at 37°C for 45 h. The remaining fraction after dissolution was used to determine the extent of crosslinking in the samples. To perform the tests, approximately 10 mg of each sample was immersed in 10 mL of deionized water and maintained at a constant temperature of 37°C using a temperature-controlled water bath and a magnetic stirrer to ensure uniform mixing. After 45 h, the undissolved fraction was collected by filtration, dried at 50°C, and weighed. The crosslinking degree was calculated as the percentage of mass retained after the dissolution process.
Determination of antimicrobial activity
The inhibition assay was carried out by the disc diffusion method against Gram-positive (Listeria monocytogenes ATCC 19,114 and Staphylococcus aureus ATCC 33,862) and Gram-negative (Escherichia coli ATCC 25,922 and Pseudomonas aeruginosa ATCC 27,853) bacteria from the official American Type Culture Collection. All indicator strains were grown overnight. Except P. aeruginosa ATCC 27,853 reactivated at 25°C in nutrient broth (NB), the other strains were cultivated at temperature of 37°C in brain heart infusion (BHI) broth. Growth media were provided by Oxoid (Milan, Italy).
The antimicrobial test was conducted with a double agar layer constituted of a 2% water agar bottom layer and a soft agar (0.7% w/v) top layer prepared with the optimal growth medium for each indicator strain inoculated at 107 CFU/ml. To achieve this, the cells were centrifuged at 5000×g for 5 min, washed twice with a physiological solution (0.85% w/v NaCl) and re-suspended in the same solution until an optical density at 600 nm (OD600) of approximately 1.00 was reached, as measured with a 6400 Spectrophotometer (Jenway Ltd, Felsted, Dunmow, UK). This OD600 value roughly corresponds to a concentration of 109 CFU/mL, as determined by plate count. The cell suspensions were then diluted 1:100, and 35 μL were added to test tubes containing soft agar media.
PVA-based membranes (6 mm diameter) were put onto the top agar layer in direct contact with the indicator bacteria. Positive control discs (6 mm diameter) were obtained from Whatman No.
1 filter soaked in 2.5% (w/v) streptomycin solution. The radial diffusion of the active substances was allowed at refrigeration temperature for 2 h before bacterial growth occurred for 24 h at the optimal temperature. The tests were conducted in duplicate (two technical replicates) and repeated twice (two independent repetitions) after a 24-h interval. A clear area around the discs indicated the inhibition and the results were expressed in mm of diameter of the halos.
Results and discussion
The fibrous structure, obtained through processing techniques that involve the use of polymer solutions, is strongly influenced by the rheological properties of the starting solutions.27,50–52 Therefore, it is crucial to examine viscosity values and rheological behavior of pure and nanoparticles drugs containing systems. Figure 2 shows the values of complex viscosity as a function of the frequency for PVA, PVA/ GNP, PVA/CHX and PVA/CHX-GNP solutions. The rheological behavior is almost the same for all investigated systems. In particular, a pseudoplastic behavior is observed with a Newtonian response in the low-frequency region, while shear thinning is shown in the higher frequency region. However, some differences are observed in terms of viscosity values. In detail, pure PVA exhibits a high Newtonian viscosity value in the low-frequency region but the highest viscosity value is shown by PVA/GNP. On the other hand, the addition of CHX, in PVA/CHX results in a decrease in the Newtonian viscosity values in all frequency range. This behavior is in complete agreement with the results obtained in previous studies also for polymer/CHX systems.27,53,54 Moreover, the addition of GNP leads to an increase in viscosity values at low frequencies both in PVA/GNP and in PVA CHX GNP. Indeed, it is widely recognized in scientific literature that the incorporation of nanoparticles typically results in increase of viscosity values.55–58 Ultimately, the viscosity values of all solutions were considered compatible with the solution blow spinning process according to previous works.
53
Complex viscosity of polymer solutions used in the HASBS technique.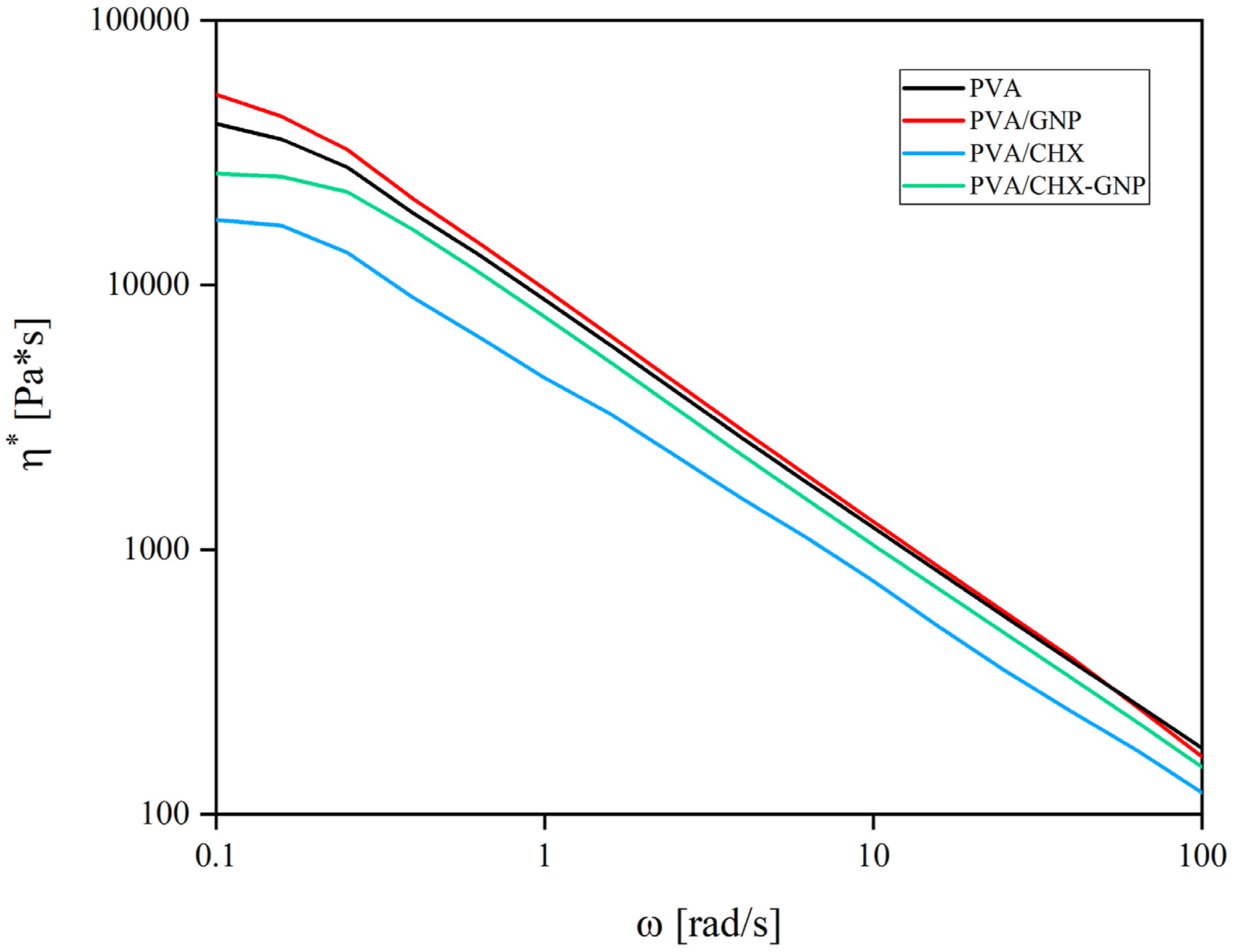
Figure 3(a)–(d) shows the SEM micrographs, including inset with the distribution diameter, of fibrous surface prepared by HA-SBS, along with their respective average diameter values. (Figure 3(e)). Neat PVA system, as shown in Figure 3(a), exhibited smooth and homogeneous fibers with unimodal diameter distribution and average diameter value ranging from 0.9 to 1.1 µm (refer to Figure 3(e)). The presence of GNP in PVA/GNP (Figure 3(b)) caused the formation of fiber bundles and an increase in the average diameter value by approximately 50%. Adding CHX to PVA (Figure 3(c)) led to the formation of homogeneous fibers with some beads and, contextually, a 20% decrease in the average diameter if compared to neat PVA. The contextual presence of drug and the nanoparticles in PVA/CHX-GNP system (Figure 3(d)) induced the formation of homogeneous and defect-free fibers. However, in this case, there was no formation of fiber bundles. SEM images of PVA fibers surfaces obtained by HASBS of (
These results are in agreement with the rheological characterizations of polymeric solutions, since it is known that low viscosity values induce the formation of fibers with a thinner diameter and vice versa.27,53,59–61
The formation of fiber bundles in the PVA GNP system could be probably attributed to the instability of the solution during the process, as observed in other previous studies.16,17,53 Furthermore, the presence of GNP played a key role in terms of increase in fibers diameter size.27,53 It is known that graphene nanoplatelets induces an increase in the viscosity values of polymer solutions.27,55,62,63 On the contrary, the presence of chlorhexidine, according to other previous work, induces a decrease in viscosity values. However, this decrease was mitigated, once again, by the presence of GNP. 27
Furthermore, it is interesting to notice that all the systems showed wavy fibers. This behavior is atypical in fibrous made by conventional SBS. The reason for the formation of wavy fibers can be probably attributed to the stabilization of PVA fibers, as already observed in other works.24,27,37,39,40 In fact, the deposition of fibers onto the heated collector probably triggered the in situ crosslinking of PVA fibers.
To better understand the stabilization mechanism a graphical description of HASBS is shown in Figure 4(a). In particular, during flight time, the evaporation of the aqueous solvent is promoted by moderately high temperature (of 60°C) inside the chamber. In fact, the production of PVA fibers, by conventional SBS technique, would be impossible at room temperatures as the aqueous solvent of the polymer solution would have difficulty to quickly evaporate during spinning. Subsequently, the PVA fibers are deposited on the hot collector, where thermal stabilization takes place, thanks to the surface temperature of the hot plate (180°C). Indeed, it is known that at this temperature, thermal crosslinking of PVA rapidly occurs.
27
Graphical description of the HASBS process (a) and thermal stabilization of PVA fibers (b).
In this regard, in order to confirm the succeed of the in situ crosslinking treatment, it was deemed appropriate to perform an FTIR-ATR analysis on the fibrous surfaces.
FTIR-ATR analysis (Figure 5 and Table 2), in addition to providing chemical and surface information of the membrane surfaces, can indirectly detect the cross-linking of fibers. In fact, cross-linking can be identified through a decrease in the OH signal (3000-3500 cm−1) owing to the dihydroxylation and oxidation reactions among the hydroxyl groups in PVA occurring during the stabilization process, providing clear evidence of the successful crosslinking of the fibers.27,64 In particular, the least intense OH signal was shown by systems containing CHX and/or GNP when compared to pure PVA. Furthermore, the examination of the C–O region (1180–1000 cm−1) clearly highlighted that the peak centered around 1150 cm−1, associated with C-O-C ether bonding, becomes increasingly prominent with the rise in crosslinking degree. ATR-FTIR spectra of PVA, CHX powder and PVA, PVA/GNP, PVA/CHX, PVA/CHX-GNP membranes surfaces. FTIR peak values and relative functional groups.
Therefore, this specific spectral region was standardized against the intensity of the C-O peak at 1090 cm−1 (as shown in the right panels), and the ratio between the signals at 1140 cm−1 and 1090 cm−1 could offer additional confirmation of the crosslinking occurrences. The system with the lowest band intensity was PVA, followed by PVA/CHX. In the opposite direction, the systems containing GNP showed the highest peak intensity. It is reasonable to think that GNP nanoparticles promoted heat transfer during the process and consequently favored thermal cross-linking, moreover, the abundance of hydroxyl groups (-OH) on the surfaces of GNP nanoparticles makes it possible for hydrogen bonds to form with hydrophilic polymers such as PVA (polyvinyl alcohol), which also has hydroxyl groups along its polymer chain. These intermolecular interactions can facilitate and accelerate the crosslinking process. 65
Furthermore, it is necessary to identify the signals attributed to the model molecule of CHX in order to confirm its potential presence within the fibrous structure. Nevertheless, the distinctive CHX peak at 1490 cm −153 was not easily identified, as reported in previously documented cases in other analog system containing CHX.
53
This is likely due to the characteristic peaks of PVA overlapping with those of CHX, especially when the latter is at low concentrations. In an attempt to confirm the actual presence, EDX analysis was conducted on solution blow spinning fibers surface to identify the potential presence of chlorine signals. The corresponding results are illustrated in Figure 6. SEM-EDX analysis verified that chlorhexidine was not only properly integrated into the fibers but also uniformly distributed across the entire surface. SEM-EDX analysis of fibers surfaces obtained by HA-SBS of PVA-CHX and PVA/CHX-GNP, Cl it is identifiable by green signal.
In particular, the EDS maps of both systems, PVA/CHX and PVA/CHX-GNP, reflect a homogeneous distribution of the Cl signal. However, in both systems, no significant variations in the intensity of the Cl signal were observed.
The crosslinking degree of the samples was determined by measuring the remaining fraction after dissolution tests carried out in deionized water at 37°C for 45 h. In fact, as reported in scientific literature, untreated PVA fibers dissolve instantly upon contact with water.22,23
Crosslinking level of PVA membranes after immersing in deionized water for 45 h at a temperature of 37°C.

These results provide strong evidence of the solid relationship between the formulation, process, and structure of the systems studied. In fact, thermal crosslinking is likely promoted thanks to the ability of graphene nanoplatelets to promote heat transfer.
27
Also, in the presence of CHX, crosslinking is promoted. This behavior could be justified by the size of the fibers. In fact, in accordance with the analysis of diameters discussed earlier, PVA-CHX fibers showed the lowest values of the mean diameter. It is reasonable that thinner fibers are more easily crosslinked as the amount and tortuous path of residual solvent is lower compared to thicker fibers.
27
The samples’ capability to promote controlled release of CHX was examined by monitoring the UV absorption wavelength at 230 nm of the CHX release at different time intervals.53,57,66,67 Figure 7 show the cumulative release of CHX expressed as Mt/M∞ ratio upon immersion time, with Mt and M∞ indicating respectively the amount of CHX released at 37°C at a given time t, and the theoretical amount of CHX integrated into the devices. Neat PVA not crosslinked (PVA/NC) was added for comparison. For both systems investigated, in contrast to the release behavior of PVA/NC fibrous systems (characterized by a single burst phase) the release of CHX is characterized by three phases. A burst phase in the initial part of the release, a second phase characterized by slower release rate and the attainment of a final plateau after long immersion times. The results show that all the mats immersed in distilled water released more than 65% of the total CHX within the first 5 h of immersion. Moreover, the plateau region is reached after 40–45 h, varying depending on the specific system. Specifically, PVA/CHX showed a more aggressive burst release, with approximately 80% of CHX released within the first 5 h. PVA/CHX-GNP, on the other hand, released approximately 60% of CHX within the first 5 h and showed a sustained release in the following immersion hours. As expected, and in accordance with previous studies,
27
the incorporation of GNP in the PVA/CHX-GNP system resulted in a higher degree of crosslinking. As a result, the GNP nanoplatelets enhanced the prolonged release of the fibers membranes. Release kinetics of PVA CHX, PVA CHX GNP and PVA NC expressed as Mt/Mloaded as a function of time.
The release data were modelized using the Peppas-Korsmeyer model, which allows for the study of release kinetics through the power law equation (1).
This model allows the identification of two key parameters: the exponent n, which describes the drug release mechanism and k which depends on the system’s properties, that is a proportionality factor that describes the overall drug release rate. In particular, when n = 0.5, the release is controlled by pure Fickian diffusion, and it usually occurs when the drug moves through the matrix following a concentration gradient without significant obstacles; n < 0.5, is referred to an hindered Fickian diffusion, where physical barriers or interactions with the matrix slow down the diffusion, though the underlying mechanism remains Fickian. When n is between 0.5 and 1, the release is anomalous or non-Fickian, where both diffusion and matrix relaxation or swelling contribute to the release process. If n = 1, the release occurs at a constant rate, typical of a first-order or Case II mechanism, often associated with constant matrix erosion or swelling. Finally, when n > 1, the release becomes accelerated, which is characteristic of systems where the matrix degrades or fragments rapidly.
The fraction of CHX released from PVA/CHX and PVA/CHX-GNP as a function of time are shown in Figure 8, where the release has been modelized considering two stages to better observe the initial rapid release (burst release) and the subsequent sustained release. Logarithmic plot of the release data expressed as Mt/M∞ as function of time for PVA/CHA and PVACHX-GNP systems.
Values of slopes (n) and intercepts (k) of fitting of Peppas-Korsmeyer model power law.

In detail, in Stage I, PVA/CHX shows a fast initial release with a value of n I = 0.2417 and a constant K I = 0.8889. PVA/CHX-GNP exhibits a slightly slower initial release, with n I = 0.2190 and K I = 0.9473. In Stage II, PVA/CHX shows a slower and more sustained release, with n II = 0.0412 and K II = 0.8533, indicating a significant reduction in the drug release rate. PVA/CHX-GNP exhibits an even more sustained release, with n II = 0.0282 and K II = 0.8982. In both stages, the values of n correspond to a hindered Fickian diffusion mechanism. Although both systems exhibit a burst release in the initial phase (Stage I), a sustained release, primarily governed by hindered diffusion phenomena, was subsequently observed. In Stage II, both samples released the entire amount of CHX theoretically incorporated within approximately 50 h. The overall hindered diffusion mechanism of both stage I and II can be explained considering the presence of a fibrous structure with a surface composed of crosslinked fibers, which limit the swelling of the bulk while at the same time allowing the immediate release of the drug available on the exposed surface. Of course, the release of the drug in stage I is attributed not only to the non-crosslinked portion, which is immediately ready to dissolve, but also to the swelling of the crosslinked or partially crosslinked fibers. The combination of these two effects can therefore be considered responsible for the behaviour of devices. Furthermore, the high amount of chlorhexidine (CHX) released in the first hour from both fibrous systems primarily comes from the non-crosslinked or partially crosslinked fibers. This behavior, already observed in other studies, is characteristic of systems with varying levels of crosslinking.27,68 In particular, during the SBS process, according to the literature and EDX analysis, the drug tends to deposit mainly on the surface of the fibers.16,53 It is therefore reasonable to hypothesize that the drug migrates from areas with a high level of crosslinking to areas with lower or absent crosslinking. This migration phenomenon is attributed to the drug’s preference for regions where the macromolecules are more mobile, typical of the non-crosslinked portions.68,69
To evaluate the durability of the device and the effectiveness of the crosslinking heat treatment, a morphological analysis of the post-release surfaces of PVA, PVA/CHX, PVA/GNP, and PVA/CHX-GNP was performed, and the results are shown in Figure 9. After 45 h of immersion, surprisingly, all the systems showed good retention of the fibrous structure, contrary to what happens with PVA/NC that rapidly dissolve when in contact with water due to the abundancy of hydroxyl functional groups (−OH) on the polymer backbone massively interact with water by which hydrogen bonds thus leading to a complete dissolution.
27
In Figure 9(a), PVA exhibited a fibrous structure with simultaneous gel formation on the surface of the fibers. PVA/GNP and PVA/CHX, respectively shown in Figure 9(b) and (c), exhibited good retention of the fibrous structure. In fact, there was no significant gel formation observed on the surface of the fibers. However, there were phenomena of fiber cohesion observed. PVA/CHX-GNP (Figure 9(d)) exhibited the best retention of the fibrous structure. In particular, it is possible to observe the presence of pores typical of fibrous membranes and a lower formation of cohesive fibers. These results are in full agreement with the crosslinking level (see Table 3), indeed, the retention of the fibrous structure in all the systems attests to the success of crosslinking and, at the same time, the ability of graphene nanoplatelets to promote the crosslinking process. Moreover, the wettability of the membrane surface was not evaluated. Unlike other studies that use long thermal stabilization processes combined with the use of cross-linking agents, where a reduction in surface wettability is observed after several hours,27,70–72 the short thermal stabilization times of HASBS (15 min) are not sufficient to penetrate bulk membranes, despite managing to stabilize a good portion of the surface. Consequently, the wettability of the membranes remains the same for both sides as that of untreated membranes, as shown in the videos added to the supporting information. To understand the influence of processing on the mechanical properties of the resulting membranes, tensile tests were conducted. The values of the elastic modulus (E), tensile strength (TS) and elongation at break (EB) for the studied systems can be found in Table 5. SEM images of surface of (a) PVA, (b) PVA/GNP, (c) PVA/CHX, (d) PVA/CHX-GNP membrane after immersion in DI water for 45 h. [E], [TS] and [EB] values of PVA, PVA/CHX, PVA/GNP and PVA/CHX-GNP fibers.

Neat PVA exhibits elastic modulus, tensile strength and elongation at break of 52.5 MPa, 4.2 MPa and 31.7% respectively. There are no significant variations in the mechanical performance for the PVA/CHX system, except for a slight decrease, likely attributable to the reduction in the average fiber diameter as observed through image analysis.
Generally, thermal cross-linking induces, as expected, an increase in E and TS values and a contextual decrease in EB values. In fact, as expected, in the presence of graphene nanoplatelets, which are capable of favoring thermal cross-linking, an increase in E and TS and a contextual decrease in EB values are observed for all systems containing GNP. Furthermore, the increase in E and TS may also be partially due to the molecular interaction between PVA and the functional groups of the GNP, thus contributing to the improved mechanical properties of the system.27,65
Inhibitory activity of PVA-based fibers.

Results indicate mean values ±standard deviation (SD) of four determinations (carried out in two technical repeats for two independent tests).
Strain codes: ATCC 25,922, Escherichia coli; ATCC 27,853, Pseudomonas aeruginosa; ATCC 19,114 Listeria monocytogenes; ATCC 33,862, Staphylococcus aureus. Positive control: 2.5% (w/v) streptomycin solution.
Conclusions
In this study, a new configuration of SBS, namely HA-SBS, was developed. Starting from a green solution, composed of distilled water and PVA, it was possible to achieve, in one-step, the production and simultaneous crosslinking of PVA fibers. The relationships among materials, processes, properties, and structure of the resulting membranes were examined through rheological, mechanical, morphological, and surface characterizations. The results revealed that all systems processed via HA-SBS exhibited satisfactory structural properties and good retention of the fibrous structure after immersion in aqueous media, contrary to non-crosslinked PVA fibrous systems that completely and rapidly dissolve upon contact with water. Furthermore, with the incorporation of GNP nanofillers, it was possible to improve the degree of crosslinking, and consequently, enhance the mechanical performance. Application release tests were conducted by incorporating the model molecule CHX into the PVA fibrous matrix. The release kinetics results showed that the HA-SBS setup mitigated the burst release typical of non-crosslinked PVA fibrous membranes. Additionally, the presence of GNP nanofillers, capable of promoting thermal cross-linking during the process, played a key role in exerting greater control over the release kinetics. Moreover, Antimicrobial tests revealed that all CHX-containing systems showed good inhibitory activity against four bacterial strains. Finally, the activation of PVA with GNP and CHX has proven its antimicrobial potential.
The one-step method of thermal in situ cross-linking enables the production of stable membranes with enhanced mechanical properties, making them suitable for various applications in biomedical, pharmaceutical, and environmental fields. Additionally, it significantly reduces processing times and eliminates the need for potentially toxic chemical cross-linking agents. Furthermore, this approach can be applied to low-volatile solvent that cannot be processed at room temperature using solution blow spinning. Further investigations could focus on optimizing the concentration of GNP nanofillers to achieve an optimal balance between enhanced mechanical properties and controlled release kinetics. This could involve a systematic study of GNP loading to identify the threshold for maximum benefits. Moreover, Investigating the scalability of the HA-SBS setup and addressing potential challenges associated with large-scale manufacturing would be valuable for future implementation.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the SAMOTHRACE (Sicilian micro and nano technology research and innovation center) Extended Partnership and received funding from the European Union Next-GenerationEU (PIANO NAZIONALE DI RIPRESA E RESILIENZA (PNRR) – MISSIONE 4 COMPONENTE 2, INVESTIMENTO 1.5) European Commission (ECS_00000022).