
Abstract
Herein, ciprofloxacin (CIP)-loaded alginate/poly (vinyl alcohol)/gelatin (SPG) (CIP–SPG) nanofiber mats were successfully fabricated by electrospinning. The average fiber diameters of the mats before and after crosslinking were in the range of 190–260 and 385–484 nm, respectively. The chemical integrity of CIP remained intact after encapsulation into the mats. The degree of weight loss and water swelling decreased with an increase in the gelatin content of the electrospun nanofiber mats. A release study was carried out by total immersion and diffusion methods using phosphate buffer as a release medium. Burst release of CIP was observed in case of the total immersion method, while a more sustained release was observed in case of the diffusion method. The maximum amounts of CIP released during total immersion and diffusion were in the range of 70–90% and 72–85%, respectively. For both the total immersion and diffusion methods, the released amounts of CIP decreased and the release slowed down with an increase in the gelatin content; this result is consistent with the weight loss and water swelling values. The Young’s modulus increased, while the tensile strength and strain at break decreased with an increase in the gelatin content. The CIP–SPG nanofiber mats were slightly toxic to L929 mouse fibroblasts as evaluated by indirect cytotoxicity assay. The electrospun CIP–SPG nanofiber mats exhibited excellent antimicrobial activity against Staphylococcus aureus and Escherichia coli. These results reveal that the electrospun CIP–SPG nanofiber mats are potentially promising materials for wound healing applications.
Introduction
Skin, as the largest organ in the human body, is responsible for protecting the body against pathogens and severe environments. Injured skin, which can be classified as acute or chronic wounds, undergoes the following four phases of wound healing: hemostasis, inflammation, proliferation, and tissue remodeling [1]. The performance of wound dressings depends on the type of matrix materials used and the added bioactive substances. Biocompatible, non-toxic, non-allergic, and biodegradable materials are suitable for use as wound dressings. The physical properties, for example, gas permeability, water uptake or swelling, and mechanical properties, of a wound dressing also affect its performance [2,3]. In addition to the selection of appropriate materials and bioactive substances, the structure of wound dressing is an important aspect. Recently, electrospun nanofibers have attracted attention for wound healing applications. Electrospinning is a process of producing ultrafine fibers in the sub-micron to nanometer range by applying a high voltage to either polymer solutions or polymer melts. Charges accumulate on the surface of a pendant drop of solution, which change the shape of the droplet to a pointed cone (known as the Taylor cone). When the electrostatic field overcomes the critical value, the cone-shaped droplet is elongated into jets that travel to the metal collector. Electrostatic and Coulombic repulsion forces originating from the accumulated charges are responsible for the stretching of the polymer jets, while the surface tension of the solution and viscoelastic force resist the elongation of these jets. The morphology and size of the electrospun nanofibers are governed by processing parameters (for example, applied voltage, tip-to-collector distance, and flow rate [4,5], solution parameters (such as viscosity, electrical conductivity, and surface tension [5], and ambient parameters (including temperature and humidity) [6].
Owing to their high surface area-to-volume ratio, the electrospun nanofibers can provide controlled and sustained release of bioactive substances compared to regular cast films [7]. Moreover, the porous structure formed owing to the non-woven texture of the randomly oriented electrospun nanofibers allows excellent water uptake, which can absorb fluids from injured tissues and offer a moist environment for tissue regeneration. Therefore, studies on the encapsulation of drugs or bioactive compounds in electrospun nanofibers as controlled release carriers were performed. Antibacterial drug-loaded electrospun polyvinylidene fluoride (PVDF) fibers were fabricated, and their mechanical properties and release behaviors were studied [8]. The antibacterial drugs were sustainably released from the PVDF fibers for three days. Dexpanthenol, an analog of vitamin B5, was loaded onto an electrospun poly (vinyl alcohol) (PVA)/sodium alginate (SA)/chitosan blend for use as a wound dressing [9], and the release profiles and kinetics of release of dexpanthenol were analyzed. Vancomycin was encapsulated in the electrospun SA/poly (ethylene oxide) (PEO)/soy protein-blended fibers, which was sustainably released from these fibers for two days [10].
Hydrogels have been widely used as wound dressing materials because of their ability to retain moisture for wound healing. SA is a derivative of alginic acid, which is a linear copolymer of (1,4)-linked β-
Furthermore, several proteins or partially hydrolyzed protein molecules, for example, gelatin, elastin, and fibroin, were incorporated into the wound dressing to enhance cell proliferation and the biocompatibility of the dressing. Alginate incorporated with an elastin-like polypeptide showed a faster rate of curcumin release and higher antioxidant activity with an increase in the elastin-like polypeptide content [17]. The blend films of strontium-loaded SA and silk fibroin showed potential for use as wound dressings because of their excellent cytocompatibility and proangiogenic action [18]. The proliferation and viability of cells increased with an increase in the gelatin content of alginate/gelatin scaffolds [19]. Among these polypeptide substances, gelatin was chosen in the present study for incorporation into electrospun nanofiber mats because of its biocompatibility, biodegradability, non-toxicity, and structural resemblance to collagen, an extracellular matrix component of tissues [20,21].
Moreover, ciprofloxacin (CIP), an antibiotic, was used as a model drug in this study. CIP is a broad-spectrum quinolone antibiotic against both Gram-negative and Gram-positive pathogens [22,23]. CIP is widely used for the treatment of various bacterial infections in humans and animals. In a number of studies reported on wound healing applications, CIP has been employed as an antimicrobial agent. The encapsulation of CIP in electrospun polyurethane/dextran [24], alginate/poly (lactic-co-glycolic acid) (PLGA) [25], and poly (
Herein, CIP-loaded alginate/PVA/gelatin (SPG) (CIP–SPG) nanofiber mats were fabricated by electrospinning, and their potential for use in wound healing applications was analyzed. The morphology, degree of weight loss and water swelling, mechanical properties, and cytotoxicity of the CIP–SPG nanofiber mats were examined. The retention of the chemical integrity of CIP after encapsulation was confirmed by proton nuclear magnetic resonance (1H-NMR) spectroscopy. The release behavior of CIP was investigated by total immersion and diffusion methods in a phosphate buffer release medium (pH 7.4) at 37 °C to simulate the physiological conditions. The antimicrobial activity of the CIP–SPG nanofiber mats was evaluated by agar disc diffusion and by percentage of bacterial reduction.
Experimental
Materials
PVA (degree of hydrolysis: 86.0–89.0%, MW: 85,000–124,000 g/mol) and gelatin powder were purchased from SD Fine Chemicals (India). SA, CIP (98%), and glutaraldehyde (GTA, 25% solution in H2O) were obtained from Acros Organics (USA). Calcium chloride (CaCl2) was purchased from Sigma-Aldrich (USA). Anhydrous disodium hydrogen orthophosphate (Na2HPO4) and sodium dihydrogen orthophosphate monohydrate (NaH2PO4.H2O) were obtained from Carlo Erba Reagents (Italy). All chemicals were of analytical grade and used without further purification.
Fabrication of electrospun SPG and CIP–SPG nanofiber mats
Electrospinning of the SPG and CIP–SPG nanofiber mats
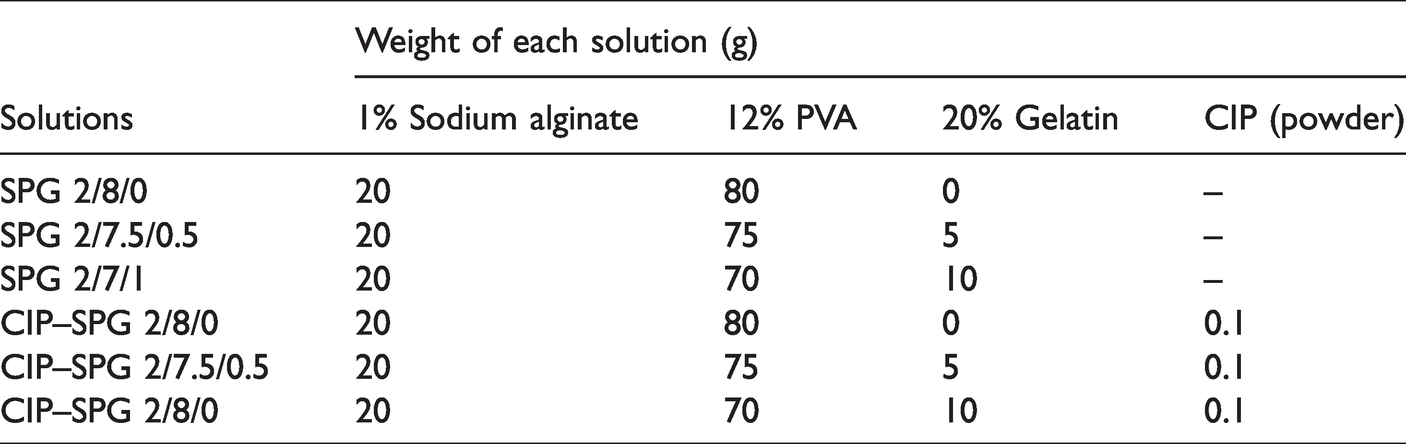
Aqueous solutions of 1% w/v SA, 12% w/v PVA, and 20% w/v gelatin were prepared at room temperature, 80°C, and 45°C, respectively. These polymer solutions were mixed at different weight ratios, as shown in Table 1. The obtained solutions were designated as SPG 2/8/0, SPG 2/7.5/0.5, and SPG 2/7/1. To achieve drug-loaded solutions, CIP was added to the abovementioned mixed polymer solutions at 0.1% w/w. The solutions were thoroughly stirred until homogeneous solutions were obtained. The resulting solutions were designated as CIP–SPG 2/8/0, CIP–SPG 2/7.5/0.5, and CIP–SPG 2/7/1. Kinematic viscosities of the as-prepared solutions were characterized using a Cannon-Fenske Routine viscometer (size number: 450 with a constant kinematic viscosity of 2.351 cSt/s at 40°C). Electrical conductivities of the as-prepared solutions were determined by a Lutron CD-4318SD conductivity meter at 25°C. Triplicate measurements of viscosity and conductivity were performed, and the average and standard deviation values were calculated.
Amounts of solutions used to prepare 100 g of mixed solutions for electrospinning.
For electrospinning, the as-prepared solution was contained in a plastic syringe connected to a stainless-steel needle used as a nozzle. The diameter of the nozzle was 0.91 mm. A high voltage of 15 kV was applied to the solutions using a Gamma High‐Voltage Research ES30P‐5W power supply. A rotating collector with a width and diameter of 25 and 7.6 cm, respectively, which was wrapped with aluminum foil was used to collect the nanofiber mats at 100 rpm. The distance between the nozzle and collector was 15 cm. The feed rate of the solution was controlled at approximately 0.5 mL/h via an SP-8800 syringe pump (AMPall, Korea). The electrospun nanofiber mats were collected for 10 h, and consequently, nanofiber mats with an average thickness of 60 ± 10 µm were achieved.
Crosslinking of the electrospun CIP–SPG nanofiber mats
The obtained CIP–SPG nanofiber mats were crosslinked using GTA vapor treatment followed by dipping in CaCl2. The CIP–SPG nanofiber mats were placed in a box containing a cup of GTA (25% in water) at 40°C for either 12 or 24 h under a saturated GTA atmosphere. The mats were placed in a hood for 1 h to allow evaporation of the remaining GTA from these mats. Subsequently, the mats were dipped in a 2% w/v CaCl2 solution for 1 s. Finally, the resulting mats were placed in a hood for 24 h and stored in a desiccator.
Characterization of the electrospun SPG and CIP–SPG nanofiber mats
Morphology of the electrospun SPG and CIP–SPG nanofiber mats
The electrospun SPG and CIP–SPG nanofiber mats before and after crosslinking were coated with a thin layer of gold using the Polaron SC7640 coater. The morphology of the samples was examined by the JSM-5410LV JEOL scanning electron microscope. The diameters of the samples were measured from scanning electron microscopy (SEM) images using the ImageJ software (version 1.52) [27]. The average diameter of the samples and standard deviation were calculated from at least 100 measurements.
Chemical compositions of the electrospun SPG and CIP–SPG nanofiber mats
The Frontier™ Spectrum 3 (Perkin Elmer) Fourier transform infrared spectrometer was used to inspect the chemical compositions of the non-crosslinked and the crosslinked nanofiber mats. The universal attenuated total reflectance (UATR) mode was utilized for both powder and liquid samples. The chemical integrity of CIP after encapsulation into the electrospun nanofiber mats was analyzed using the Ascend™ 600 (Bruker) 1H-NMR spectrometer. SPG 2/7/1, CIP–SPG 2/7/1, and pristine CIP were dissolved in deuterated dimethyl sulfoxide (DMSO-d6), and their 1H-NMR spectra were obtained at a resonance frequency of 600 MHz.
Weight loss and water swelling behaviors of the electrospun CIP–SPG nanofiber mats
Water swelling and weight loss behaviors of the electrospun CIP–SPG nanofiber mats were investigated by diffusion using Franz diffusion cells. Each mat was placed and clamped on top of the Franz diffusion cell filled with phosphate buffer (pH 7.4) and maintained at 37°C. The Franz diffusion cell had a diameter and volume of 13 mm and 9 mL, respectively. The initial dry weight of the mat was recorded as Mi. At 2 and 4 h of diffusion, the wet mat was detached from the Franz diffusion cell, and its weight was recorded as M. Subsequently, the mat was dried in an oven at 40°C for 12 h. The dry weight after diffusion was recorded as Md. The degree of weight loss of the mat was calculated using equation (1)
The degree of water swelling is usually calculated according to equation (2)
The mats had a significantly high degree of weight loss, which led to imprecise water swelling values because some mass of the mats was lost during their diffusion (or submersion in other cases) in the medium. Therefore, equation (3) was employed to calculate modified water swelling by eliminating the factor of weight loss from equation (2). The term Mi in equation (2) was replaced by the term Mr, as presented in equation (4), which represented the weight of the sample that remained after subtracting the weight loss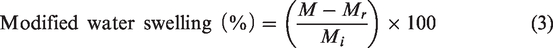

The measurement of weight loss and modified water swelling were performed in triplicate.
Release behavior of CIP from the electrospun CIP–SPG nanofiber mats
Encapsulation efficiency of CIP
Prior to studying the release characteristics of CIP from the electrospun CIP–SPG nanofiber mats, the encapsulation efficiency of CIP in the nanofibers was determined for being used as base values in release study. Each specimen was cut into squares of 2 × 2 cm2 and its weight was recorded. The specimen was immersed in a mixed solution of 15 mL phosphate buffer (pH 7.4) and 15 mL dimethyl sulfoxide (DMSO). The solution was stirred until the specimen was completely dissolved. The amount of CIP in the solution was quantified by the I3 Hanon UV–vis spectrophotometer at a wavelength of 270 nm. The absorbance was obtained and calculated into the amount of CIP by utilizing a pre-determined calibration curve of CIP in phosphate buffer/DMSO solution. The encapsulation efficiency of CIP in the nanofiber mat was calculated as percentage of the actual amount divided by the loaded amount of CIP. The experiments were performed in triplicate.
CIP-release assay
The release characteristics of CIP from the electrospun CIP–SPG nanofiber mats were investigated by the total immersion and diffusion methods. For the total immersion method, each CIP–SPG nanofiber mat was cut into squares of 2 × 2 cm2 followed by immersion in 40 mL phosphate buffer (pH 7.4) at 37°C. Then, the phosphate buffer was slowly stirred continuously during the release time ranging from 0 to 48 h. For the diffusion method, each CIP–SPG nanofiber mat was placed and clamped on top of the Franz diffusion cell filled with phosphate buffer. Water at a controlled temperature of 37°C was circulated in the Franz diffusion cells. At specified time points ranging from 0 to 48 h, 1.0 and 0.3 mL release media were withdrawn from the total immersion and diffusion systems, respectively, and their absorbance was measured by the I3 Hanon UV–vis spectrophotometer at a wavelength of 270 nm. Fresh phosphate buffer in the abovementioned amounts was refilled in the respective release bottles. The amount of CIP released from the samples was evaluated against the predetermined standard curve of CIP concentration in phosphate buffer and absorbance. The cumulative percentage of CIP release was calculated according to equation (5)
Mechanical properties of the electrospun CIP–SPG fiber mats
The mechanical properties, including tensile strength, Young’s modulus, and strain at break, of the electrospun CIP–SPG nanofiber mats were analyzed by the NRI Narin Instrument (Thailand) Universal Testing Machine according to the guidelines outlined by American Standard Testing Methods (ASTM-D882). The mats with an average thickness of approximately 60 ± 10 µm were cut into rectangles of 10 × 90 mm2 that were tested under a load of 10 kN at 25°C. The gauge length and crosshead speed were 50 mm and 10 mm/min, respectively. For each type of mat, at least five measurements were carried out.
Cytotoxicity of the electrospun CIP–SPG fiber mats
Indirect cytotoxicity of the electrospun CIP–SPG nanofiber mats was determined based on the ISO 10993-5 standard using L929 mouse fibroblasts (ATCC CCL1, NCTC 929, and strain L). The cells were cultured in minimum essential medium (MEM) at a density of 1 × 105 cells/mL in a 96-well tissue culture polystyrene plate followed by incubation at 37°C under a 5% CO2 and 95% humid atmosphere for 24 h. Furthermore, the mats were cut into squares of 2 × 2 cm2 followed by sterilization using UV irradiation for 15 min on each side. Each fiber mat was immersed in MEM at 37°C for 24 h at a surface area-to-volume extraction ratio of 3 cm2/mL. These extraction media were used to evaluate the cytotoxicity of the mats. The media of the cell culture were removed and replaced by the media extracted from the tested samples in which the cells were further incubated for 24 h. The negative control (NC) and positive control (PC) were the media extracted from a Thermanox (Nunc) plate and from a polyurethane film containing 0.1% zinc diethyldithiocarbamate, respectively. Fresh culture medium was used as a blank. All the media extracted from the mats, NC, and PC were prepared at a similar surface area-to-volume extraction ratio of 3 cm2/mL. Finally, the viability of the cells was assessed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay. The details of the MTT assay are provided in the Supplementary Information (S1). A microplate reader (EASYS, UVM240 S/N ASY54180) was used to measure the optical density of the purple formazan solution obtained from the MTT assay at a wavelength of 570 nm, which was proportional to the number of living cells. The percentage of cell viability was calculated according to equation (6)
Antibacterial activity of the electrospun CIP–SPG fiber mats
Antibacterial activities of the electrospun CIP–SPG nanofiber mats against Staphylococcus aureus (ATCC 25,923) and Escherichia coli (ATCC 25,922) were evaluated by two methods, namely agar disc diffusion and percentage of bacterial reduction. The size of the tested square mats was 50 × 50 mm2. The mats were sterilized by UV irradiation for 60 min on each side before testing. The experiments were performed in triplicate.
Agar disc diffusion
The antibacterial activity of the mats was determined by the agar disc diffusion method based on the Japanese Industrial Standard (JIS L1902) for woven and non-woven textiles. The mat was placed on a glass plate (diameter: 100 mm) containing bacteria in agar. The NC and PC were filter papers saturated with distilled water and ethanol, respectively. The plate was then incubated at 37°C for 18 h. The length of the inhibition zone (the area without bacteria) was computed from the edge of the sample to the end of the clear zone using the ImageJ software (version 1.52) [27].
Percentage of bacterial reduction
The percentage of bacterial reduction was evaluated according to the Japanese Industrial Standard (JIS Z 2801:2006), which is used for testing the antimicrobial activity of plastics and adapted from ISO 22196:2011. The colony-forming units (CFUs) of bacteria after treatment for 24 h were compared with the initial CFUs. Each mat was inoculated with 0.4 mL of approximately 105 CFU/mL bacteria followed by incubation in the dark at 37°C under a 90% humid atmosphere for 24 h. The percentage of bacterial reduction was determined according to equation (7)
Antibacterial effectiveness of the mats was assessed by the antibacterial activity (R) according to equation (8)
Statistical analysis
All data were presented as mean ± standard deviation. Statistical analysis in the studies of weight loss, modified water swelling, mechanical properties, cytotoxicity, and agar disc diffusion was carried out by using a paired two sample for means: t-test using the Microsoft Excel software. The statistical difference between two sets of data was considered when p < 0.05.
Results and discussion
Fabrication of the electrospun SPG and CIP–SPG nanofiber mats
Electrospinning of the SPG and CIP–SPG nanofiber mats
Electrospun nanofiber mats were successfully fabricated from the blends of SA, PVA, and gelatin and designated as SPG 2/8/0, SPG 2/7.5/0.5, and SPG 2/7/1 based on the amounts of polymer solutions used to prepare 100 g of mixed solutions for electrospinning, as described in Table 1. For the encapsulation of CIP in the mats, CIP powders were added to 0.1% w/w SPG solutions before electrospinning. The CIP–SPG nanofiber mats were designated as CIP–SPG 2/8/0, CIP–SPG 2/7.5/0.5, and CIP–SPG 2/7/1. Figure 1 shows the selected SEM images of the electrospun SPG and CIP–SPG nanofiber mats obtained at different blending ratios. Round nanofiber mats with smooth surfaces were obtained under all conditions. The encapsulation of CIP did not alter the surface morphology of these mats. The fiber diameters of the mats were measured from the SEM images, and their distribution curves have been presented along with the SEM images. The average fiber diameters of the mats were calculated and have been listed in Table 2. The average fiber diameters of the electrospun SPG and CIP–SPG nanofiber mats were in the range of 193–234 and 190–262 nm, respectively.

SEM images and distribution curves of the fiber diameters of (a) SPG 2/8/0, (b) SPG 2/7.5/0.5, (c) SPG 2/7/1, (d) CIP–SPG 2/8/0, (e) CIP–SPG 2/7.5/0.5, and (f) CIP–SPG 2/7/1 (magnification: 7500 with a scale bar of 1 µm).
Viscosity and electrical conductivity of the as-prepared solutions and average fiber diameters of the electrospun SPG and CIP-loaded SPG nanofiber mats.

The average fiber diameters for both the SPG and CIP–SPG nanofiber mats decreased with an increase in the gelatin content. The fiber diameters were affected by the processing, solution, and ambient parameters. Kinematic viscosity and electrical conductivity of the as-prepared solutions were investigated before electrospinning and have been summarized in Table 2. With an increase in the gelatin content, the viscosities of both the SPG and CIP–SPG solutions decreased. The decrease in the viscosity of the solutions led to a smaller viscoelastic force, which is a force that resists the stretching of the polymer jet because of electrostatic and Coulombic repulsion forces. Therefore, the elongation of the electrospun nanofibers was enhanced; in other words, the fiber diameter was smaller when the viscosity of the solution was lower [28]. Electrical conductivity is another important parameter that affects the diameter of electrospun nanofibers. With an increase in the gelatin content, the electrical conductivities of both the SPG and CIP–SPG solutions increased. Gelatin, also referred to as hydrolyzed collagen [29], with a high dipole moment arising from the amino acid functional group of proteins, may cause a high electrical conductivity of the solution. Consequently, the higher electrical conductivity of the solution leads to higher electrostatic and Coulombic repulsion forces, which result in the thinning down or elongation of the fiber jets [5]. Therefore, the fiber diameters decreased with an increase in the gelatin content due to the decrease in viscosity and increase in the electrical conductivity of the solutions.
Crosslinking of the electrospun CIP–SPG fiber mats
For wound dressing applications, the mechanical properties of materials are also important. Crosslinking of matrix molecules is usually performed to improve mechanical properties of materials. A number of crosslinking agents containing carboxylic group, e.g., maleic acid, isocyanate group, and aldehyde group, e.g., GTA are used to crosslink PVA and gelatin. Among various kinds of crosslinking agents, GTA is widely used according to its accessibility and inexpensiveness. Successful crosslinking of PVA [30,31] and gelatin [32,33] by using GTA were reported, whereas the crosslinking of alginate was performed by using CaCl2 [34]. However, in order to minimize the toxicity of GTA that might be remained in the samples, a vapor treatment method was performed instead of direct immersion to reduce the contact between samples and GTA. The electrospun CIP–SPG nanofiber mats were crosslinked using GTA vapor for either 12 or 24 h. The SEM images of the crosslinked nanofiber mats are presented in Table 3.
SEM images and average fiber diameters (ϕ) of the crosslinked electrospun CIP-loaded SPG nanofiber mats (magnification: 7,500 with a scale bar of 1 µm).

After 12 h of GTA crosslinking, the electrospun CIP–SPG nanofiber mats fused together with the presence of junction points. The average fiber diameters of the GTA-crosslinked CIP–SPG 2/8/0 and CIP–SPG 2/7/1 were 385 ± 25 and 484 ± 25 nm, respectively. Interestingly, although the non-crosslinked CIP–SPG 2/7/1 was smaller (190 ± 40 nm) than the non-crosslinked CIP-SPG 2/8/0 (262 ± 60 nm), the crosslinked CIP–SPG 2/7/1 had a larger fiber diameter than the crosslinked CIP–SPG 2/8/0. The initial smaller fiber diameter of CIP–SPG 2/7/1, which possesses higher surface area, led to a higher degree of fiber fusion, resulting in a larger diameter of CIP–SPG 2/7/1 after crosslinking. Upon further crosslinking with GTA vapor for 24 h, most of the fiber mats fused together, and they exhibited a film-like morphology rather than a fiber-like morphology. Therefore, the GTA vapor treatment for 24 h was unsuitable for crosslinking the CIP–SPG nanofiber mats. An attempt to crosslink alginate molecules was performed after crosslinking PVA and gelatin using GTA vapor for 12 h. The CIP–SPG nanofiber mats were rapidly dipped in a CaCl2 solution. Most of the mats dissolved or fused together and became transparent films. This result suggested that crosslinking along with the retention of the nanofiber mat morphology could be achieved only with the 12-h GTA vapor treatment. For further studies such as those on mechanical properties, chemical integrity, the release of CIP, cytotoxicity, and antimicrobial activity, only the CIP–SPG nanofiber mats crosslinked via the 12-h GTA vapor treatment were used.
Chemical compositions of the electrospun SPG and CIP–SPG nanofiber mats
Fourier-transform infrared (FT-IR) spectroscopy
The electrospun CIP–SPG nanofibers were crosslinked with GTA vapor before use in the release study. It is therefore questionable whether GTA would be remained in the nanofibers. To examine that the non-crosslinked and the crosslinked electrospun CIP–SPG 2/8/0 and 2/7/1 nanofiber mats were investigated by FT-IR spectroscopy. Aqueous solution of GTA which was used in the crosslinking process was also examined. For GTA, the peaks at about 3338 and 1640 cm−1 corresponded to O–H stretching from hydrogen bonding and C = O stretching, respectively (see Figure 2). For the non-crosslinked and the crosslinked electrospun CIP-SPG 2/8/0 and 2/7/1 nanofiber mats, their spectra appeared to resemble. The peaks at about 3317, 2940, and 1732 cm−1 corresponded to O–H stretching, C–H stretching, and C = O stretching, respectively. The peaks belong to C–H bending and C–O stretching appeared at about 1245 and 1091 cm−1, respectively. Even though, the peaks of C = O was observed in the crosslinked electrospun CIP–SPG specimens, this peak appeared at the different wave number from that of GTA. This evidence determined that there was no GTA remained in the specimens.

FT-IR spectra of the non-crosslinked and the crosslinked electrospun CIP–SPG 2/8/0 and CIP–SPG 2/7/1 and glutaraldehyde aqueous solution.
Proton-nuclear magnetic resonance (1H-NMR) spectroscopy
Chemical integrity of CIP encapsulated in the electrospun SPG nanofiber mats was examined by 1H-NMR spectroscopy. Figure 3 shows the spectra of the electrospun SPG nanofiber mats without CIP, CIP powder, and electrospun CIP–SPG nanofiber mats. In the spectrum of electrospun SPG nanofiber mats, the peaks of alginate, PVA, and gelatin were observed in the range of 0.9–5.2 ppm. In the spectrum of CIP powders, the peaks corresponding to the different protons at positions a, aʹ, b, bʹ, c, d, e, f, g, gʹ, h, and hʹ (see the chemical structure beside the spectrum) were obtained in the range of 1.2–8.7 ppm. The other peaks at 2.5 and 3.3 ppm belonged to DMSO (used as a solvent) and residual moisture, respectively. In the spectrum of electrospun CIP–SPG nanofiber mats, the peaks corresponding to CIP and SPG were retained, thereby indicating that the chemical integrity of CIP remained intact even under high electrostatic forces during electrospinning.

1H‐nuclear magnetic resonance spectra of the crosslinked electrospun SPG 2/7/1 nanofiber mat, CIP powder, and crosslinked CIP–SPG 2/7/1.
Weight loss and modified water swelling of the electrospun CIP–SPG nanofiber mats
Data on the weight loss and modified water swelling of the electrospun CIP–SPG nanofiber mats are summarized in Figure 4(a) and (b), respectively. For all types of samples, the weight loss and modified water swelling values increased with an increase in diffusion time. The weight loss values of CIP–SPG 2/8/0, CIP–SPG 2/7.5/0.5, and CIP–SPG 2/7/1 after 2 h of diffusion were 48% ± 1%, 11% ± 2%, and 11% ± 2%, whereas those after 4 h of diffusion were 60% ± 9%, 33% ± 11%, and 31% ± 7%, respectively. Upon comparing the weight loss values of the abovementioned samples after 2 and 4 h of diffusion, it was found that the values of CIP–SPG 2/7.5/0.5 and CIP–SPG 2/7/1 were not significantly different. However, the weight loss of CIP–SPG 2/8/0 was significantly higher than that of the other samples (p < 0.05).

Graphs of the (a) weight loss and (b) modified water swelling of the crosslinked electrospun CIP-loaded SPG nanofiber mats. *Significant at p < 0.05 with respect to CIP–SPG 2/8/0 for each time of diffusion.
The modified water swelling values for CIP–SPG 2/8/0, CIP–SPG 2/7.5/0.5, and CIP–SPG 2/7/1 after 2 h of diffusion were 466% ± 136%, 466% ± 48%, and 467% ± 37%, whereas those after 4 h of diffusion were 749% ± 253%, 638% ± 50%, and 638% ± 52%, respectively. These values after 2 h of diffusion were not significantly different among all types of samples. However, the value after 4 h of diffusion of CIP–SPG 2/8/0 was significantly higher than that of the other samples (p < 0.05). The degree of swelling of the nanofiber mats did seem considerably high but not much as presented in other works [35,36]. The alginate/gelatin scaffolds crosslinked by CaCl2 for use in soft tissue engineering exhibited a degree of swelling of about 800% in Hank’s balanced salt solution (HBSS) at 37°C [35]. The degree of swelling of the alginate-based hydrogels crosslinked with CaCl2 immersed in tap water for 4 h at 25°C were about 33 gram of water/gram of dry hydrogel which were equivalent to 3300% [36].
In this study, both the weight loss and the modified water swelling decreased with an increase in the gelatin content. The decrease in water swelling and weight loss with the addition of gelatin was consistent with the corresponding trends reported in a previous study [37] in which with the addition of gelatin, PVA/poly(ethylene glycol) (PVA/PEG) hydrogels exhibited less water swelling than that exhibited by neat PVA/PEG hydrogels because of the higher crystalline content. The materials that can absorb and retain more water contribute to a higher degree of degradation or higher weight loss because water can penetrate the matrix. In other words, the materials with higher degree of water swelling, which implied that they were more swollen and more surrounded by media molecules, exhibited the higher degree of weight loss. Weight loss and modified water swelling behaviors are important factors for describing and controlling the release characteristics of substances [38–40] and therefore have been discussed further along with the release behaviors in the next section.
Release behaviors of CIP from the electrospun CIP–SPG nanofiber mats
Encapsulation efficiency of CIP
Prior to studying the release characteristics of CIP from the electrospun CIP–SPG nanofiber mats, the encapsulation efficiency of CIP in the nanofiber mats was determined. The encapsulation efficiency of CIP in the electrospun CIP–SPG 2/8/0, CIP–SPG 2/7.5/0.5, and CIP–SPG 2/7/1 were 91.17% ± 6.65%, 88.58% ± 3.13%, and 87.41% ± 9.38%, respectively. These values were used to calculate the actual amounts of CIP in the nanofiber mats and subsequently being used as base values in release study to calculate the percentage of CIP released.
Release of CIP
The release behaviors of CIP from the electrospun CIP–SPG nanofiber mats were investigated by the total immersion and diffusion methods using the Franz diffusion cell. The release study was carried out in phosphate buffer (pH 7.4) at 37°C to simulate the physiological pH and temperature conditions. The release amounts of CIP were calculated against the predetermined standard curve of CIP in phosphate buffer (pH 7.4) revealed in the Supplementary Information (S2). The release profile of CIP from the electrospun CIP–SPG nanofiber mats obtained using the total immersion method is shown in Figure 5(a). The cumulative amounts of CIP released from the samples were reported as the percentages of the weights of CIP released divided by the initial weights of CIP in the samples. The release of CIP was observed in the immersion time range of 0–2,880 min (48 h). For all types of mats, burst release of CIP was observed at the initial time of immersion until about 45 min followed by a sustained release. After approximately 180 min (3 h), the cumulative release of CIP reached a plateau. The maximum cumulative release of CIP from CIP–SPG 2/8/0, CIP–SPG 2/7.5/0.5, and CIP–SPG 2/7/1 was approximately 98% ± 3%, 91% ± 6%, and 81% ± 8%, respectively. Apparently, the release of CIP reduced with an increase in the gelatin content of the mats. This result is in agreement with the weight loss and modified water swelling behaviors. Similar to the cases of weight loss and water swelling, the amount of CIP released from CIP–SPG 2/8/0 was greater than that released from CIP–SPG 2/7.5/0.5 and CIP–SPG 2/7/1. When the matrix is highly solvated by the medium (that is, when there is more water swelling), the drug molecules conveniently diffuse out of the matrix [40,41]. Additionally, based on the weight loss behavior of the matrix, the drug molecules can be released during the erosion of the matrix [41].

Cumulative release of CIP from the crosslinked electrospun CIP–SPG nanofiber mats by the (a) total immersion method and (b) diffusion method using the Franz diffusion cell.
Nevertheless, the total immersion method is not a perfect model to describe the actual release characteristics of substances. The observations of this method can be used to preliminarily distinguish between the release behaviors of different wound dressing materials in the medium. The diffusion method is more suitable as it is easy to simulate the physiological release conditions for this method. The release profile of CIP from the electrospun CIP–SPG nanofiber mats obtained by the diffusion method using a Franz diffusion cell is illustrated in Figure 5(b). A sustained release of CIP was achieved for all types of mats. The cumulative release of CIP reached a plateau at approximately 1,440 min (24 h), which was significantly slower than that in the case of the total immersion method. The maximum cumulative release of CIP from CIP–SPG 2/8/0, CIP–SPG 2/7.5/0.5, and CIP–SPG 2/7/1 was approximately 93% ± 6%, 82% ± 6%, and 79% ± 4%, respectively. Similar to the results of the total immersion method, the release of CIP lowered with an increase in the gelatin content of the mats. The amount of CIP released was consistent with the amounts of weight loss and water swelling, as mentioned above.
Mechanical properties of the electrospun CIP–SPG nanofiber mats
Ideal wound dressing materials should be biocompatible, non-toxic, non-allergic, antimicrobial, and should maintain a moist environment and promote cell migration and proliferation [3,42]. In addition to the abovementioned aspects, the wound dressing materials should have acceptable mechanical properties. The tensile strength, Young’s modulus, and strain at break of the electrospun CIP–SPG nanofiber mats are illustrated in Figure 6. The tensile strengths of the electrospun SPG nanofiber mats significantly decreased (p < 0.05) with an increase in the gelatin content, which were 4.01 ± 0.21, 3.55 ± 0.36, and 2.92 ± 0.70 MPa for SPG 2/8/0, SPG 2/7.5/0.5, and SPG 2/7/1, respectively. The Young’s modulus significantly increased (p < 0.05) with an increase in the gelatin content; values were 16.0 ± 2.0, 29.4 ± 7.7, and 48.4 ± 4.7 MPa for SPG 2/8/0, SPG 2/7.5/0.5, and SPG 2/7/1, respectively. With the addition of gelatin, the strain at break of SPG 2/7.5/0.5 (55% ± 8%) remarkably decreased as compared to that of SPG 2/8/0 (117% ± 29%). However, the strains at break of SPG 2/7.5/0.5 and SPG 2/7/1 (48% ± 11%) were not substantially different. Apparently, with the addition of gelatin, the electrospun nanofiber mats exhibited greater stiffness and less elasticity. This result is similar to those reported in the literature [43]; the tensile strength and elongation at break of the electrospun PVA/gelatin fibers decreased with an increase in the gelatin content. Additionally, the Young’s modulus of the PVA/PEG hydrogels improved with the addition of gelatin because of the increase in crystallinity [37].

Mechanical properties of the crosslinked electrospun SPG and CIP–SPG nanofiber mats: (a) tensile strength, (b) Young’s modulus, and (c) strain at break. *Significant at p < 0.05 with respect to SPG 2/8/0. #Significant at p < 0.05 with respect to SPG 2/7.5/0.5. ΔSignificant at p < 0.05 with respect to CIP–SPG 2/8/0. □Significant at p < 0.05 with respect to CIP–SPG 2/7.5/0.5.
The effects of gelatin addition on the tensile strength, Young’s modulus, and strain at break of the electrospun CIP–SPG nanofiber mats showed the same trends as those in the case of the SPG nanofiber mats. The tensile strengths of CIP–SPG 2/8/0, CIP–SPG 2/7.5/0.5, and CIP–SPG 2/7/1 were 3.64 ± 0.53, 3.59 ± 0.50, and 3.07 ± 0.50 MPa, respectively, which were comparable to those of the SPG nanofiber mats. The Young’s modulus values of CIP–SPG 2/8/0, CIP–SPG 2/7.5/0.5, and CIP–SPG 2/7/1 were 38.6 ± 1.8, 46.1 ± 1.6, and 54.6 ± 8.8 MPa, respectively, which were greater than those of the SPG nanofiber mats with the same content of polymer matrix. The Young’s modulus increased significantly (p < 0.05) with increasing gelatin content. The values of strain at break of CIP–SPG 2/8/0, CIP–SPG 2/7.5/0.5, and CIP–SPG 2/7/1 were 55% ± 16%, 51% ± 2%, and 32% ± 3%, respectively, which were lower than those of the SPG nanofiber mats. Only CIP–SPG 2/7/1 showed significant lower value of strain at break than that of the other samples (p < 0.05). It should be noted that the statistical significances presented in Figure 6 were considered either within a group of SPG samples or a group of CIP–SPG samples but not across the groups. As evidenced, after the encapsulation of CIP, the resulting electrospun nanofiber mats exhibited greater stiffness, less flexibility, and less elasticity probably because the rigid CIP particles disturbed the flexibility of the polymer chains. A number of aliphatic and aromatic rings in CIP could contribute to its rigidity that diminished mobility of polymer molecules. This is similar to the case of recycled natural rubber in which the presence of aluminum powder causes a decrease in the elongation and an increase in the hardness of the composites because the mobility of the matrix chains is restricted by the aluminum particles [44].
Cytotoxicity of the electrospun CIP–SPG nanofiber mats
Indirect cytotoxicities of CIP–SPG 2/8/0 and CIP–SPG 2/7/1 were investigated using the L929 mouse fibroblasts based on the ISO 10993-5 standard. After culturing the cells with the media extracted from the tested nanofiber mats, the percentages of viable cells were evaluated and have been illustrated in Figure 7. The cell viabilities of the blank, NC, and PC were 100.0% ± 0.0%, 99.3% ± 0.4%, and 0.0% ± 0.0%, respectively. SPG 2/7/1 with no CIP loading showed a cell viability of 83.3% ± 0.9% which was significantly lower than NC (p < 0.05). Nevertheless, this result indicated that SPG 2/7/1 was nontoxic to cells because its cell viability was greater than 70%, which is generally considered as nontoxic. However, CIP–SPG 2/8/0 and CIP–SPG 2/7/1 exhibited significant lower cell viabilities (p < 0.05) than that of SPG 2/7/1. Also, the cell viability of CIP–SPG 2/8/0 was significantly lower than the value of CIP–SPG 2/7/1. The results indicated that CIP–SPG 2/8/0 and CIP–SPG 2/7/1 appeared to be slightly toxic to cells because of their lower cell viabilities (57.3% ± 5.0% and 65.2% ± 0.9%, respectively). As the in vitro cytotoxicity testing was performed by culturing the cells with the media extracted for 24 h from the tested samples, the results might differ from those observed in the in vivo environment. Under in vivo conditions, the CIP released from the mats will be removed over time owing to the circulation of fluids in living tissues. Therefore, the actual cytotoxicity of the electrospun CIP–SPG nanofiber mats is expected to be less than that observed in the in vitro experiments.

Indirect cytotoxicity evaluation of the crosslinked electrospun CIP–SPG nanofiber mats based on the viability of L929 mouse fibroblasts. *Significant at p < 0.05 with respect to the negative control. #Significant at p < 0.05 with respect to SPG 2/7/1. ΔSignificant at p < 0.05 with respect to CIP–SPG 2/7/1.
Antibacterial activities of the electrospun CIP–SPG nanofiber mats
Agar disc diffusion
The application potential of the electrospun CIP–SPG nanofiber mats as wound dressings was evaluated by determining their antibacterial properties. The antibacterial properties of CIP–SPG 2/8/0 and CIP–SPG 2/7/1 were tested against two types of bacteria, that is, Gram-positive S. aureus and Gram-negative E. coli, by the agar disc diffusion method. Figure 8(a) shows the photographs of bacteria cultured plates. The length of the inhibition zone was measured and has been shown in Figure 8(b). Distilled water and ethanol were used as an NC and a PC, respectively. Greater lengths of the inhibition zone were observed for both CIP–SPG 2/8/0 and CIP–SPG 2/7/1, thereby indicating their excellent antibacterial effectiveness against both types of bacteria. The lengths of inhibition zones for both CIP–SPG 2/8/0 and CIP–SPG 2/7/1 were even higher than that for the ethanol (PC) (significant at p < 0.05). Upon comparing the antibacterial activities of CIP–SPG 2/8/0 and CIP–SPG 2/7/1, the length of inhibition zone for CIP–SPG 2/8/0 was found to be significantly greater than that for CIP–SPG 2/7/1 (p < 0.05). This result is in accordance with the abovementioned release results of CIP. The higher release of CIP from CIP–SPG 2/8/0 could contribute to higher antibacterial property.

(a) Photographs of bacteria cultured plates and (b) lengths of the inhibition zone for the crosslinked electrospun CIP–SPG nanofiber mats, a negative control (NC), and a positive control (PC) against S. aureus and E. coli. *Significant at p < 0.05 with respect to ethanol (PC). #Significant at p < 0.05 with respect to CIP–SPG 2/7/1.
Percentage of bacterial reduction
The antibacterial properties of CIP–SPG 2/8/0 and CIP–SPG 2/7/1 were further evaluated by the percentage of bacterial reduction. The CFUs before and after the inoculation of CIP–SPG 2/8/0 and CIP–SPG 2/7/1 with either S. aureus or E. coli. were measured and have been summarized in Table 4. Evidently, the reduction of both S. aureus and E. coli after a 24-h treatment with both CIP–SPG 2/8/0 and CIP–SPG 2/7/1 was 100%. These results revealed the excellent antibacterial properties of these mats. The R values were subsequently determined and listed in Table 5. Both CIP–SPG 2/8/0 and CIP–SPG 2/7/1 had the R values of 3.28 and 6.26 against S. aureus and E. coli, respectively. As the R is greater than 2, the tested samples qualify as effective antimicrobial materials. Noticeably, based on the agar disc diffusion and bacterial reduction results, both CIP–SPG 2/8/0 and CIP–SPG 2/7/1 exhibited excellent antibacterial properties, which could highlight them as good candidates for biomedical applications, for example, wound healing and transdermal drug delivery.
Percentage of bacterial reduction for the crosslinked electrospun CIP–SPG nanofiber mats based on investigations in S. aureus and E. coli.

Antibacterial activity (R) of the crosslinked electrospun CIP–SPG nanofiber mats based on investigations in S. aureus and E. coli.

Conclusions
In the present study, encapsulation of CIP into the electrospun SPG nanofibers was achieved. The fiber diameters decreased with an increase in the gelatin content owing to the decrease in the viscosity and increase in the electrical conductivity of the solutions. The chemical integrity of the specimens was confirmed by FT-IR and 1H-NMR spectroscopy. The release characteristics of CIP were investigated by the total immersion and diffusion methods at 37°C with phosphate buffer (pH 7.4) as the release medium. A burst release of CIP was initially observed in case of the total immersion method. The CIP release reached a plateau at approximately 180 min, and the maximum amount of CIP released was in the range of 81–98%. For the diffusion method, a slower release was noticed. The release of CIP reached a plateau at approximately 24 h, and the maximum amount of CIP released was in the range of 79–93%. For both the abovementioned methods, the release of CIP decreased and slowed down with an increase in the gelatin content; this result was consistent with the degree of weight loss and water swelling. With an increase in the gelatin content of the electrospun CIP–SPG nanofiber mats, the stiffness of the mats increased, while the elasticity decreased. The application potential of the electrospun CIP–SPG nanofiber mats as wound dressings was examined by evaluating their cytotoxicity and antimicrobial activity. The electrospun CIP–SPG nanofiber mats exhibited excellent antimicrobial activity against S. aureus and E. coli, as determined by the agar disc diffusion and bacterial reduction assays. These results indicate that the electrospun CIP–SPG nanofiber mats are promising materials for wound dressing applications because they can sustainably release CIP due to their good mechanical properties and antimicrobial activities.
Supplemental Material
sj-pdf-1-jit-10.1177_1528083721997466 - Supplemental material for Ciprofloxacin-loaded alginate/poly (vinyl alcohol)/gelatin electrospun nanofiber mats as antibacterial wound dressings
Supplemental material, sj-pdf-1-jit-10.1177_1528083721997466 for Ciprofloxacin-loaded alginate/poly (vinyl alcohol)/gelatin electrospun nanofiber mats as antibacterial wound dressings by Tittaya Thairin and Patcharaporn Wutticharoenmongkol in Journal of Industrial Textiles
Footnotes
Acknowledgements
The authors acknowledge the support received from the Faculty of Engineering, Thammasat University.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors acknowledge the funding from the Faculty of Engineering and the research unit in polymer rheology and processing, Thammasat University.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.