
Abstract
Keywords
Introduction
Endovascular therapy of lower extremity peripheral artery disease (PAD) initially involved only balloon dilation, but stenting evolved rapidly to repair suboptimal dilation and improve long-term patency diminished by the high rates of restenosis after balloon angioplasty. However, stent implantation is challenging in very mobile arterial segments, such as in the femoropopliteal segment, and is often avoided in joint areas to prevent compression fracture or collapse of the stent. The treatment of in-stent restenosis (ISR) is also more challenging than repeat revascularization after balloon angioplasty. 1 Yet none of the mechanical therapies addresses the underlying neointimal hyperplasia resulting from vessel inflammation caused by balloon inflation or permanent stent implantation. 2
Drug-coated balloons (DCBs) were developed to overcome these limitations. Many randomized controlled trials (RCTs)3–7 have shown consistent superior outcomes of DCBs over uncoated balloons in femoropopliteal lesions, with data up to 5 years for some studies.8–11 This large body of evidence led several medical societies, including the Society for Cardiovascular Angiography and Interventions, to recommend the use of DCBs for the treatment of femoropopliteal lesions. 12 In the infrapopliteal lesions, the outcomes have been controversial so far.13,14
Passeo-18 Lux (Biotronik AG, Buelach, Switzerland) is a DCB that has been successfully tested in the BIOLUX P-I and P-II studies, where it showed superior performance outcomes to an uncoated balloon in femoropopliteal lesions 4 and comparable results in infrapopliteal lesions. 13 The aim of the BIOLUX P-III all-comers registry was to collect clinical performance data and midterm safety data on the Passeo-18 Lux in a large unselected patient population seen in daily clinical practice. Results of the first 200 patients have been published 15 and now the outcomes of the 700-patient all-comers cohort are presented.
Materials and Methods
Study Design
This prospective, nonrandomized, observational registry, which was conducted at 41 centers in 15 countries in Europe, Asia, and Australia, has been previously described. 9 The aim was to evaluate the safety and performance of the Passeo-18 Lux DCB in a large unselected patient population, including below-the-knee lesions and Rutherford category 5 or 6, under real-world conditions. Patients were eligible if they had lesions in the infrainguinal arteries suitable for endovascular treatment with the Passeo-18 Lux DCB, were at least 18 years old, and signed a patient data release form or patient informed consent form. Excluded were pregnant patients, those with a life expectancy <1 year, those having unsuccessful target lesion crossing with a guidewire. Enrolment of the first 700 patients occurred from October 2014 to February 2016 and was expanded to 877 patients treated until January 2017 to reach the minimum subgroup sizes.
The procedure, follow-up assessments, and antiplatelet therapy were at the investigators’ discretion and according to standard of care at the study centers. Data were collected at baseline/intervention, at discharge, and at 6-, 12-, and 24-month follow-up visits. The visit window was ±30 days for the 6-month and ±60 days for the 12- and 24-month visits. At the time the study was designed, 24 months were considered sufficient for midterm follow-up, but the study follow-up has now been prolonged to 5 years.
The registry was conducted according to the Declaration of Helsinki and ISO14155:2011 as applicable and approved by the regional ethics review board affiliated with each participating center. All patients provided informed consent. To ensure data quality, a risk-based monitoring approach was applied, with at least 25% randomly chosen subjects fully monitored. Besides, all target lesion revascularizations (TLR) and major adverse events (MAEs) were adjudicated by an independent clinical events committee (CEC). The trial was registered on the National Institutes of Health website ( ClinicalTrials.gov ; identifier NCT02276313).
Study Device
The Passeo-18 Lux DCB has been previously described.2,8 In brief, the balloon is coated with 3 µg paclitaxel per mm2 incorporated in a butyryl-trihexyl-citrate excipient. Sizes available during the observation period were 2.0- to 7.0-mm diameter and lengths of 40, 80, and 120 mm. Balloon diameters up to 4.0 mm are 4-F–compatible and diameters from 5.0 to 7.0 mm 5-F–compatible. The safeguard insertion aid protects the user and coating from contact and damage and reduces the drug loss due to friction within the introducer sheath. It is premounted on the balloon and does not require any preparation prior to use.
Study Population
This analysis includes the 12-month results for the all-comers cohort, which comprised the first 700 patients (mean age 70.0±10.2 years; 439 men) enrolled in the BIOLUX P-III registry and a subgroup analyses of patients with at least 1 lesion in the femoropopliteal segment (493 patients, 578 lesions) and 88 subjects with 99 ISR lesions.
The patient baseline characteristics for the entire cohort and the subgroups are summarized in Table 1. In the all-comers cohort, more than half of the patients had a history of PAD and a previous peripheral intervention; nearly half of the participants had diabetes. More than a third of subjects (234, 37.7%) had chronic limb-threatening ischemia (CLTI). Of these, 128 (26.2%) subjects had Rutherford category 5 or 6 ischemia. In both subgroups, the proportions of diabetes, CLTI, and coronary artery disease were similar to the all-comers group. The ISR subgroup had a higher percentage of patients with a smoking history and hyperlipidemia.
Baseline Patient Characteristics. a
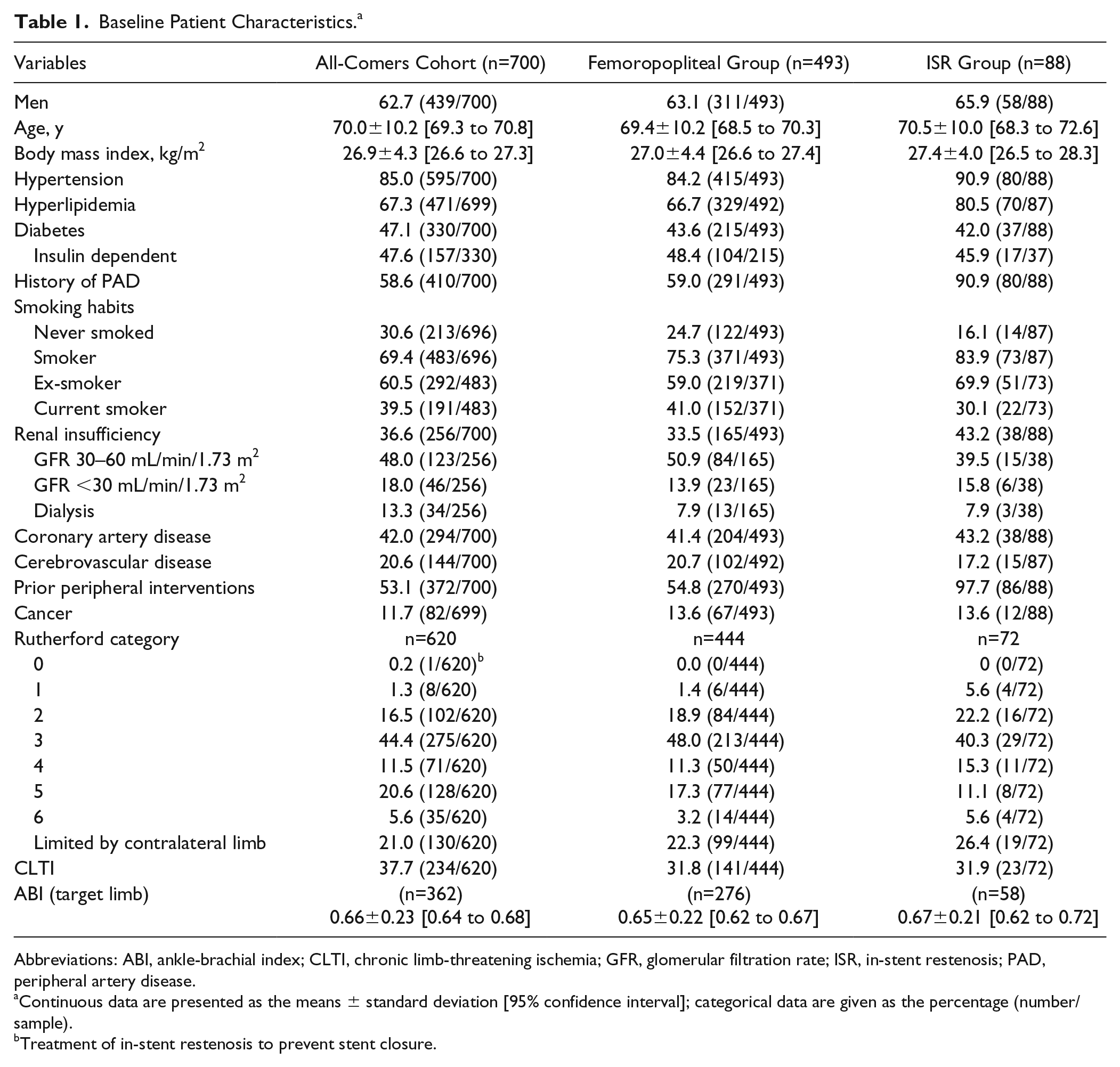
Abbreviations: ABI, ankle-brachial index; CLTI, chronic limb-threatening ischemia; GFR, glomerular filtration rate; ISR, in-stent restenosis; PAD, peripheral artery disease.
Continuous data are presented as the means ± standard deviation [95% confidence interval]; categorical data are given as the percentage (number/sample).
Treatment of in-stent restenosis to prevent stent closure.
Of 863 lesions treated (Table 2), 685 (79.4%) were in the femoropopliteal segment and 11.9% in the infrapopliteal arteries. More than half of the lesions were de novo (463, 53.7%), and 205 (23.8%) were occluded. Three quarters of lesions was calcified (42.1% moderate or heavy), and a quarter of the lesions (226/855, 26.4%) were TransAtlantic Inter-Society Consensus C or D lesions. Mean target lesion length was 84.7±73.3 mm and the mean reference vessel diameter was 4.8±1.0 mm. The lesion characteristics in the subgroups were in the same range as in the all-comers cohort. On average, the lesion length and reference vessel diameter were greater in the femoropopliteal subgroup. There were fewer calcified lesions in the ISR subgroup.
Baseline Lesion Characteristics. a
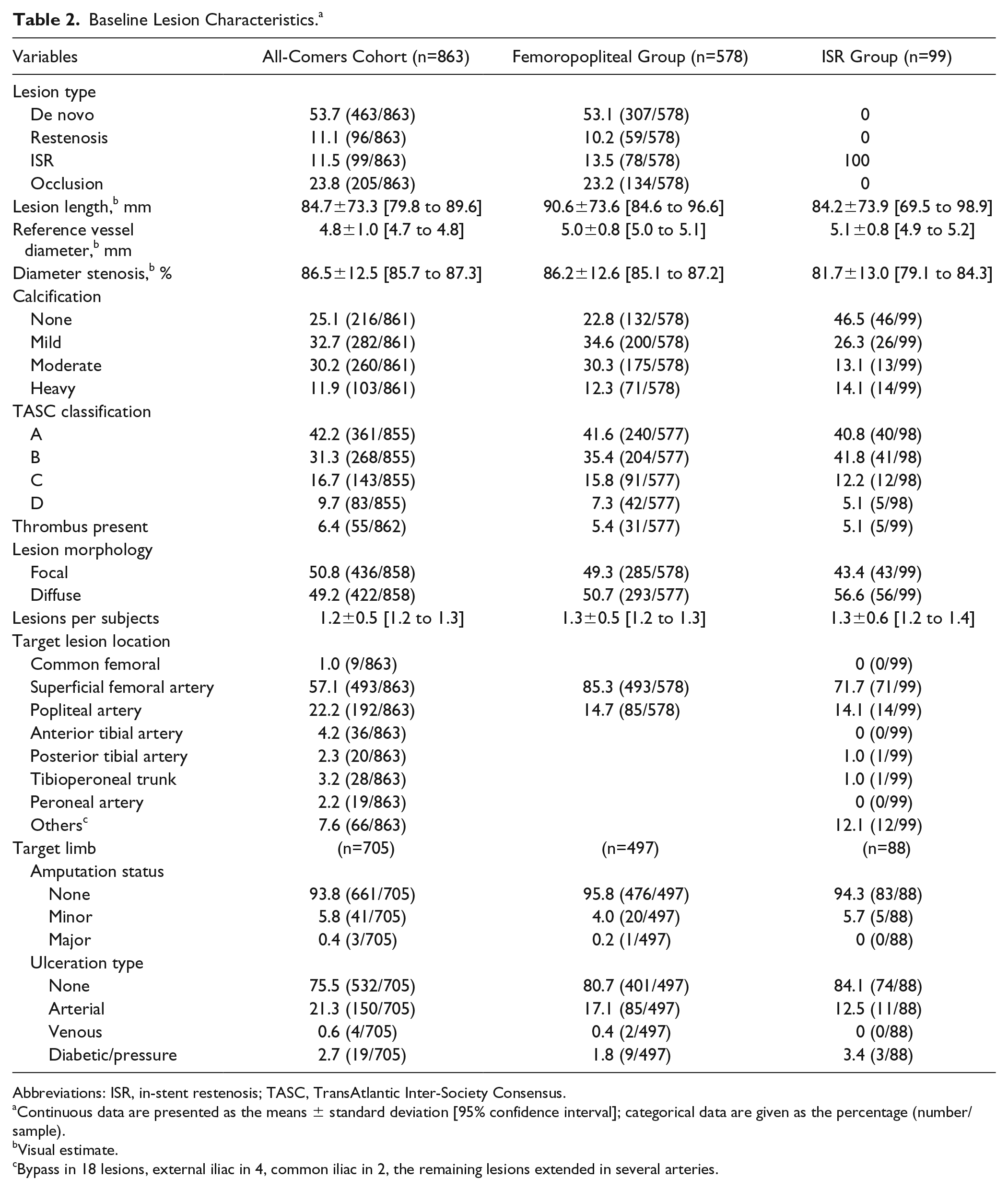
Abbreviations: ISR, in-stent restenosis; TASC, TransAtlantic Inter-Society Consensus.
Continuous data are presented as the means ± standard deviation [95% confidence interval]; categorical data are given as the percentage (number/sample).
Visual estimate.
Bypass in 18 lesions, external iliac in 4, common iliac in 2, the remaining lesions extended in several arteries.
Vessel preparation (Table 3) was performed in 625 (72.4%) cases, predominantly with a plain balloon [559 (89.4%) of the pretreated lesions], but also with cutting or scoring balloons (36, 5.7%) or atherectomy devices (16, 2.6%). In the ISR subgroup, however, the rate of vessel preparation was lower compared to the all-comers cohort and the femoropopliteal subgroup. Additional stenting was required in 145 (16.8%) of the overall 863 lesions. Not quite at third of the occluded lesions (63/205, 30.7%) were stented. In the subgroups, a bailout stent was implanted in 117 (20.2%) femoropopliteal lesions and 6 (6.1%) ISR lesions.
Procedure Characteristics. a
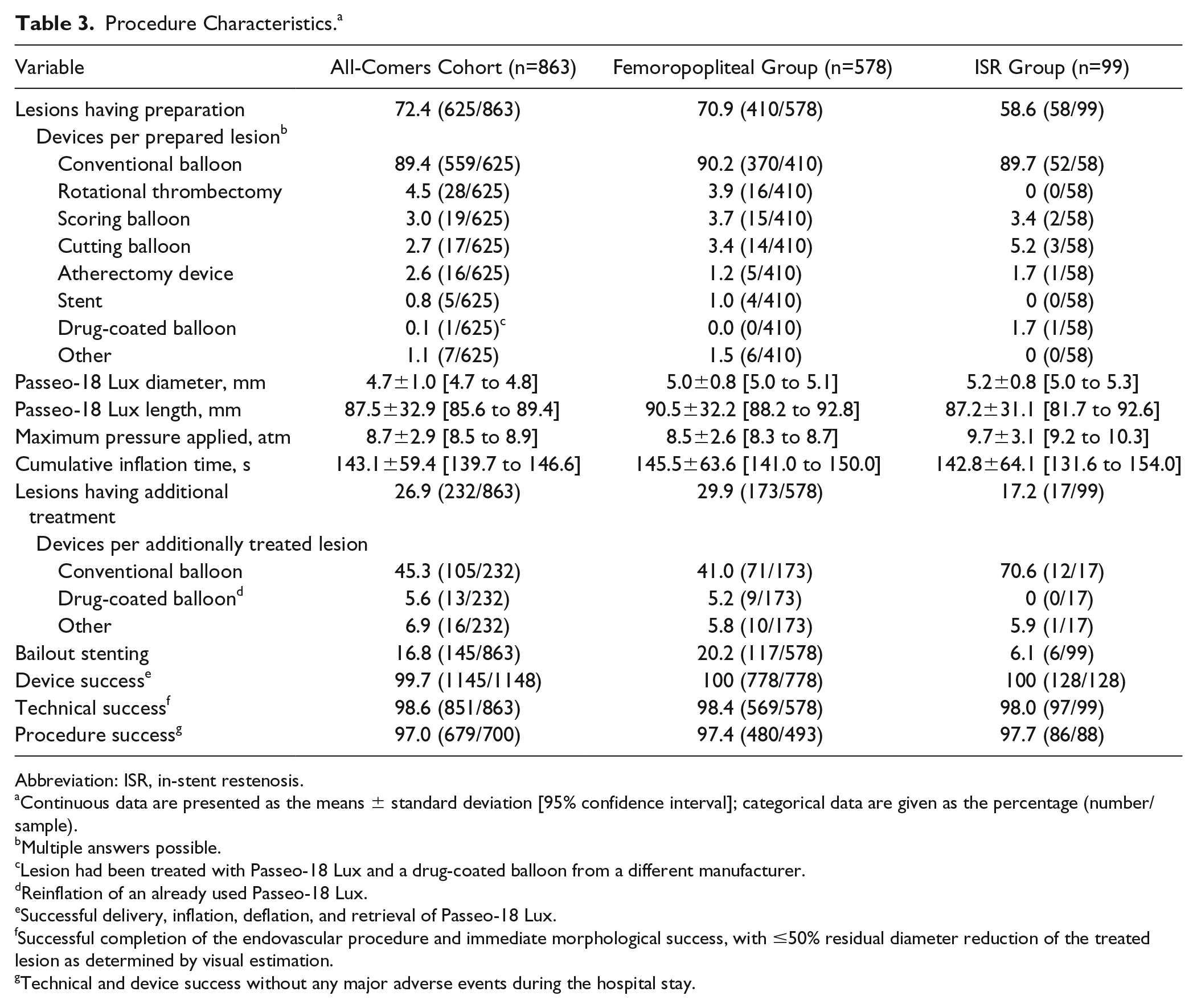
Abbreviation: ISR, in-stent restenosis.
Continuous data are presented as the means ± standard deviation [95% confidence interval]; categorical data are given as the percentage (number/sample).
Multiple answers possible.
Lesion had been treated with Passeo-18 Lux and a drug-coated balloon from a different manufacturer.
Reinflation of an already used Passeo-18 Lux.
Successful delivery, inflation, deflation, and retrieval of Passeo-18 Lux.
Successful completion of the endovascular procedure and immediate morphological success, with ≤50% residual diameter reduction of the treated lesion as determined by visual estimation.
Technical and device success without any major adverse events during the hospital stay.
Study Outcomes and Subgroup Analyses
The clinical primary endpoint was freedom from MAEs, a composite of device- and procedure-related mortality through 30 days, freedom from major target limb amputation, and freedom from clinically-driven TLR within 6 months after the procedure. The 12-month primary performance measure was freedom from clinically-driven TLR, defined as any reintervention performed for ≥50% diameter stenosis (visual estimate) at the target lesion after documentation of recurrent clinical symptoms.
Secondary endpoints were freedom from clinically-driven TLR at 6 and 24 months and at 6, 12, and 24 months freedom from clinically-driven target vessel revascularization and amputation-free survival (the latter a composite of no target limb major amputation or death). Freedom from MAEs and primary patency (defined as freedom from >50% restenosis in the target lesion as indicated by a peak systolic velocity ratio >2.5 on duplex ultrasound or by visual assessment of an angiogram with no clinically-driven reintervention) were assessed at 12 and 24 months. Primary patency and TLR were evaluated on a per lesion basis. Additionally, the ankle-brachial index (ABI) and patient-reported outcomes of the pain scale and walking impairment questionnaire were documented at each follow-up visit.
Four success rates are reported: (1) clinical success, defined as improvement in the Rutherford category at follow-up compared with the preprocedure level; (2) device success—successful delivery, inflation, deflation, and retrieval of the Passeo-18 Lux DCB; (3) technical success—successful completion of the endovascular procedure and immediate morphological success with ≤50% residual stenosis at the lesion site as determined by visual estimation; and (4) procedure success—technical and device success without any MAE during the hospital stay.
Statistical Analysis
Considering the observational registry design, BIOLUX P-III did not involve hypothesis-driven sample size estimation. The overall sample size of 700 subjects for this observational registry was determined to ensure a sufficient number of patients in the predefined subgroups. Continuous variables are presented as mean ± standard deviation; categorical variables are presented as number/sample and percentage. Comparisons to baseline variables were conducted using the t test, Wilcoxon signed-rank test, or exact sign test for paired data. Clinical outcomes were estimated using the Kaplan-Meier method; standard errors were calculated using the Greenwood formula. Estimates are reported with the 95% confidence intervals (CI). The threshold of statistical significance was p<0.05. All statistical analyses were carried out using SAS (version 9.4; SAS Institute Inc, Cary, NC, USA).
Results
Patient disposition at follow-up in the all-comers cohort is displayed in Figure 1. The primary clinical endpoint, freedom from MAEs, was 94.0% (95% CI 91.9% to 95.5%) at 6 months and 89.5% (95% CI 86.9% to 91.6%) at 12 months (Figure 2A), mainly driven by TLR. At 12 months, the primary performance endpoint (Figure 3A), freedom from clinically-driven TLR, was 93.1% (95% CI 91.1% to 94.7%).

Patient flow for the all-comers cohort.
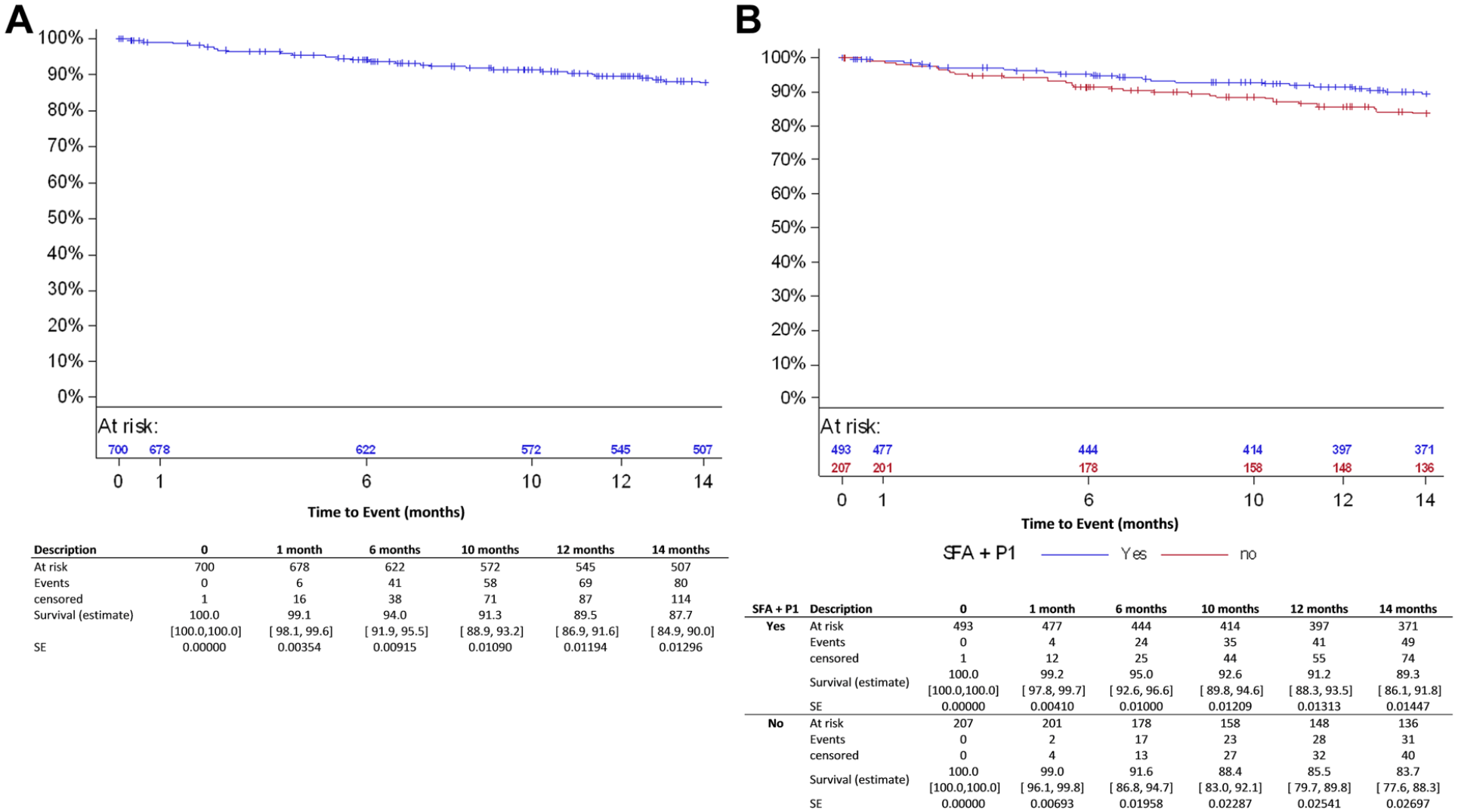
Kaplan-Meier estimates for freedom from major adverse events in (A) the all-comers cohort and (B) and the femoropopliteal group. SFA, superficial femoral artery; P1, popliteal artery segment 1; SE, standard error.
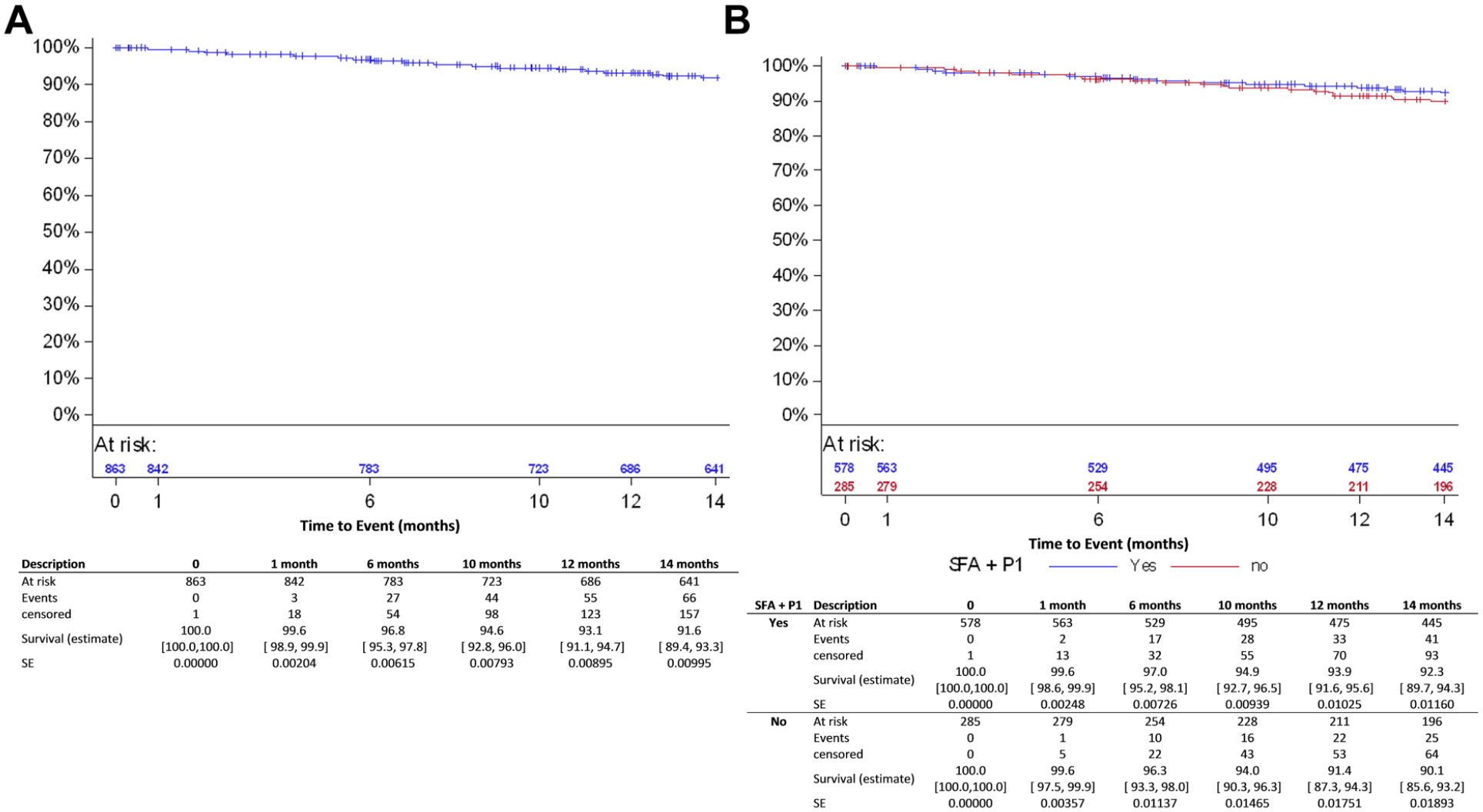
Kaplan-Meier estimates for freedom from clinically-driven target lesion revascularization (lesion based) in (A) the all-comers cohort and (B) and the femoropopliteal group. SFA, superficial femoral artery; P1, popliteal artery segment 1; SE, standard error.
The Kaplan-Meier estimate for primary patency (Figure 4A) was 85.3% (95% CI 82.6% to 87.6%) at 12 months (Table 5). However, inherent to an observational registry, duplex ultrasound assessment was not mandatory. Therefore, asymptomatic binary restenosis has not been determined for all patients. In the subgroup of patients with imaging assessment done at 12 months (299 subjects/361 lesions), the patency rate at 12 months was 79.6% (95% CI 75.1% to 83.5%).
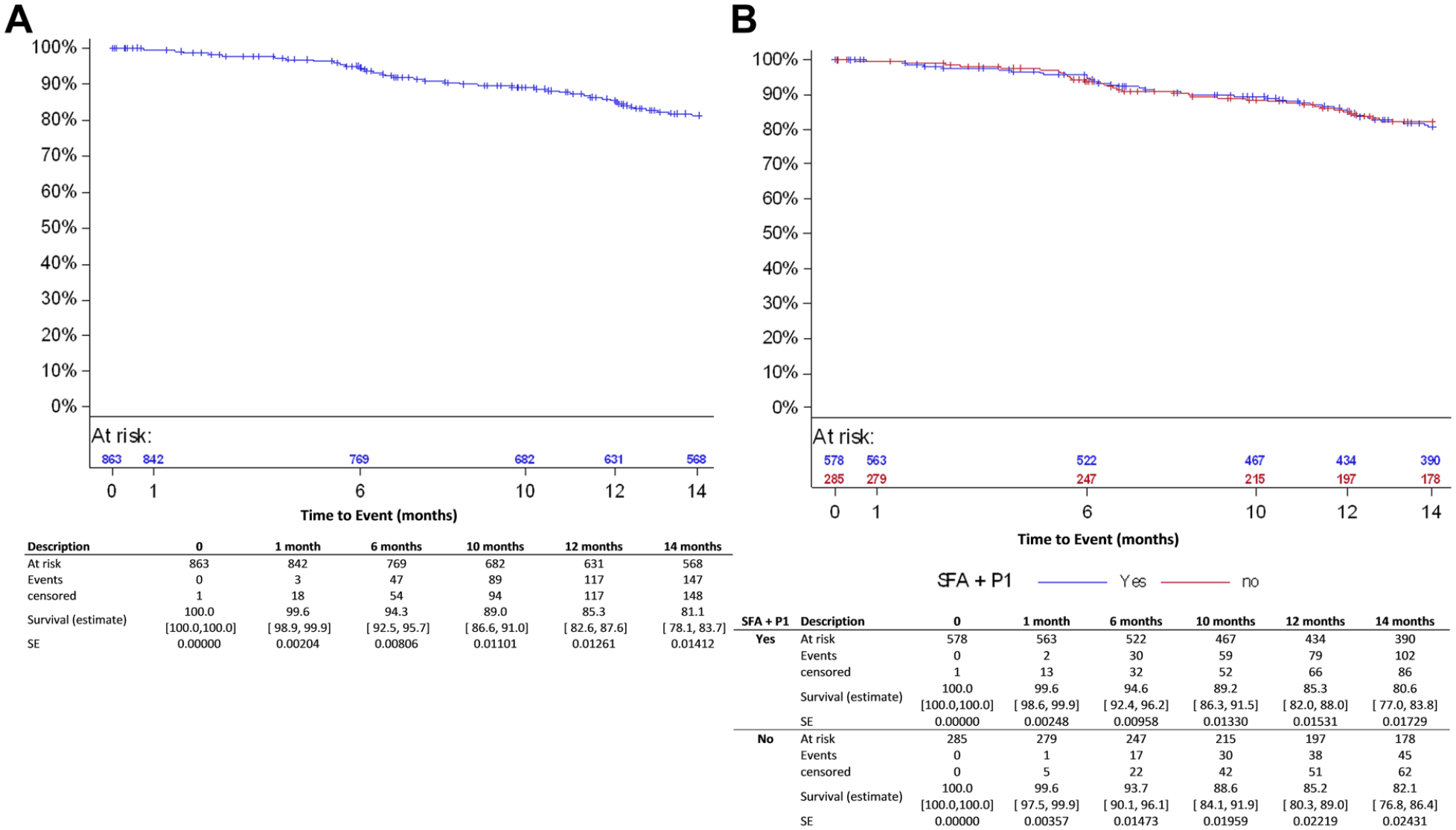
Kaplan-Meier estimates for primary patency (lesion based) in (A) the all-comers cohort and (B) and the femoropopliteal group. SFA, superficial femoral artery; P1, popliteal artery segment 1; SE, standard error.
Sixteen subjects, of whom 14 were diabetic, had a major target limb amputation, 11 had Rutherford category 5 or higher at baseline. All amputations were adjudicated by the CEC as not device related. Six patients died within 30 days after the index procedure. Through the 12-month follow-up, 41 (6.1%) patients (20 of which were Rutherford category 5 or 6) had died (Figure 5A). None of the deaths (causes are listed in Supplementary Table 1; available in the online version of the article) was considered procedure- or device-related by the CEC.
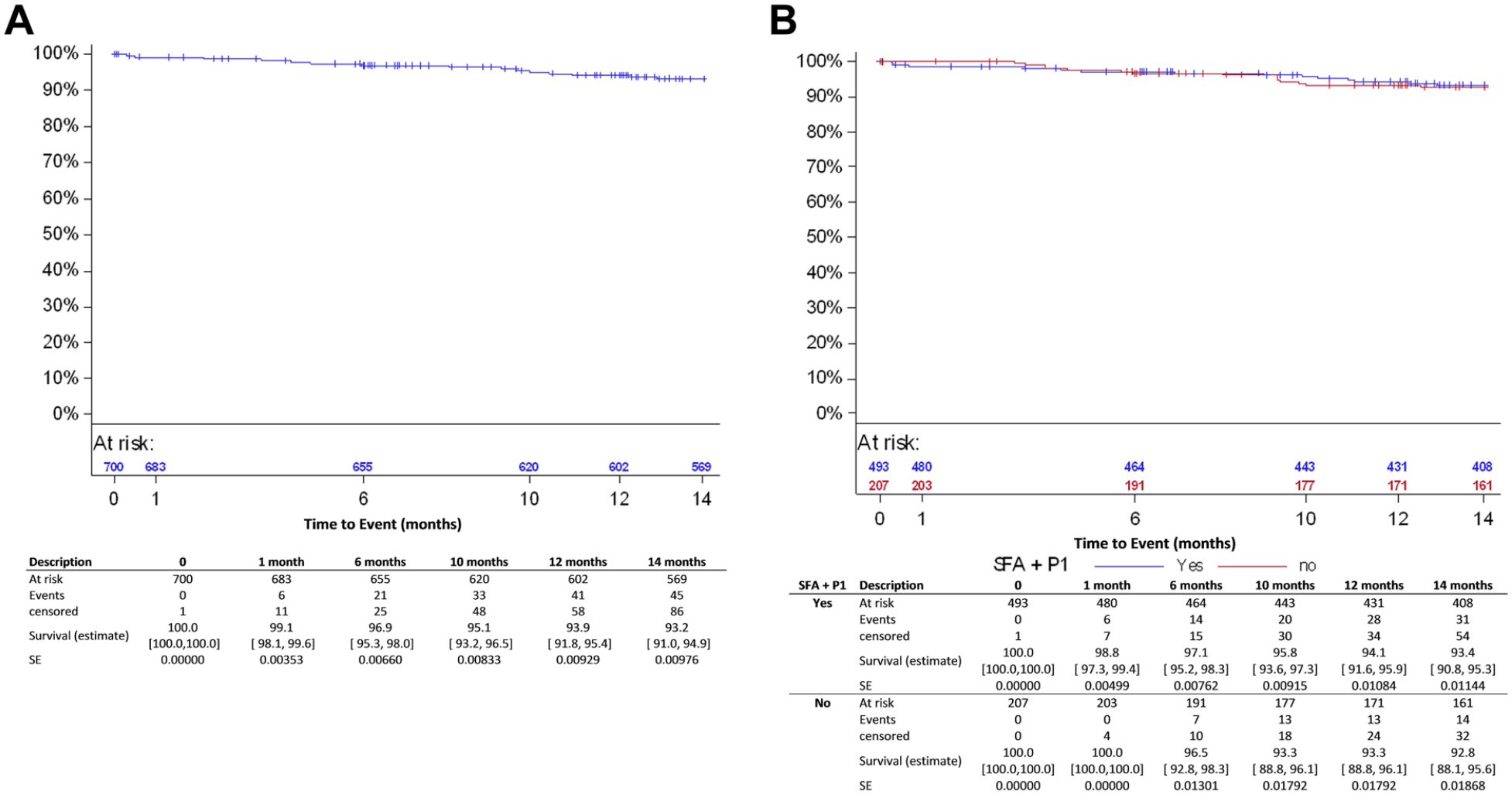
Kaplan-Meier estimates for survival in (A) the all-comers cohort and (B) and the femoropopliteal group. SFA, superficial femoral artery; P1, popliteal artery segment 1; SE, standard error.
ABI improved significantly by 0.2 from baseline at the 6- and 12-month follow-up (p<0.001; Table 4), and 310 (81.8%) of 379 patients evaluated improved at least 1 Rutherford category between baseline and the 12-month follow-up.
Hemodynamic Outcomes in Follow-up Compared With Baseline Values. a
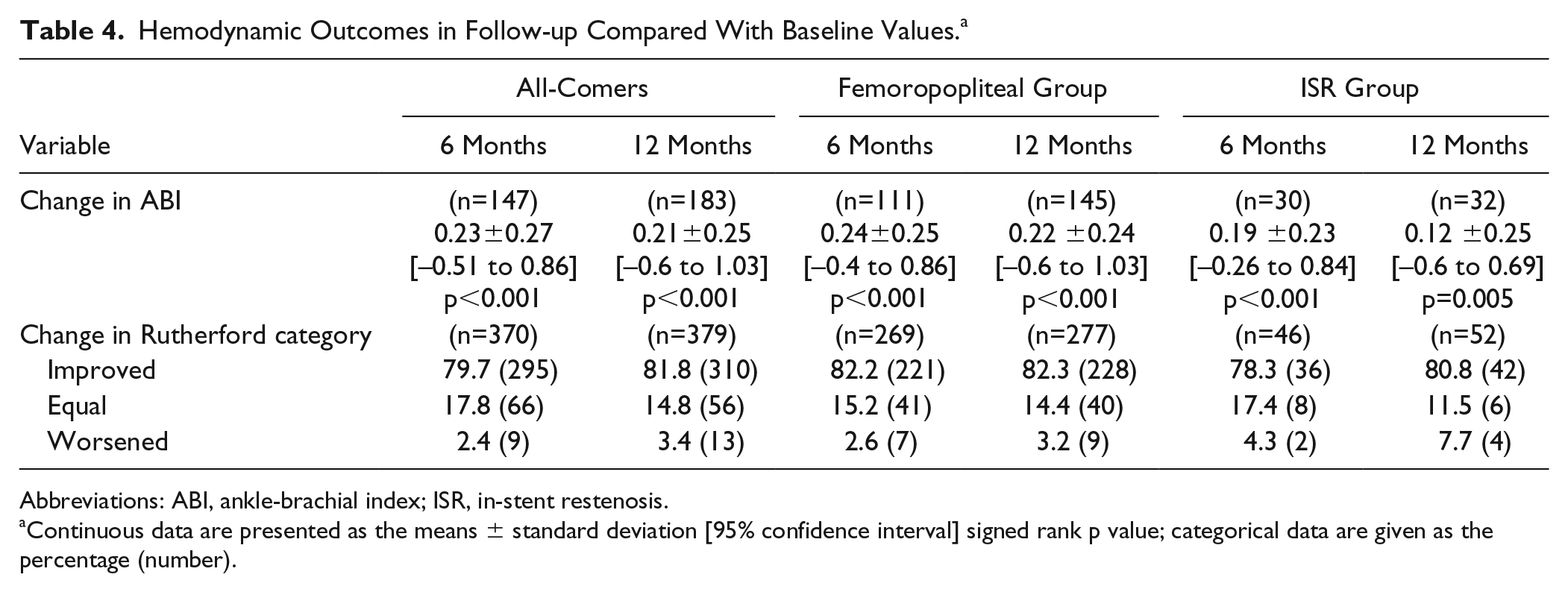
Abbreviations: ABI, ankle-brachial index; ISR, in-stent restenosis.
Continuous data are presented as the means ± standard deviation [95% confidence interval] signed rank p value; categorical data are given as the percentage (number).
Subgroup Analysis
Freedom from MAEs, the primary clinical endpoint, at 6 and 12 months was estimated as 95.0% (95% CI 92.6% to 96.6%) and 91.2% (95% CI 88.3% to 93.5%) in the femoropopliteal subgroup (Figure 2B) and 95.3% (95% CI 88.0% to 98.2%) and 88.0% (95% CI 78.9% to 93.4%) in the ISR subgroup, respectively (Table 5). The primary performance endpoint, freedom from clinically-driven TLR, was 93.9% (95% CI 91.6% to 95.6%) in the femoropopliteal subgroup (Figure 3B) and 89.4% (95% CI 81.1% to 94.1%) for ISR (Table 5).
Kaplan-Meier Estimates of Clinical and Performance Outcomes. a
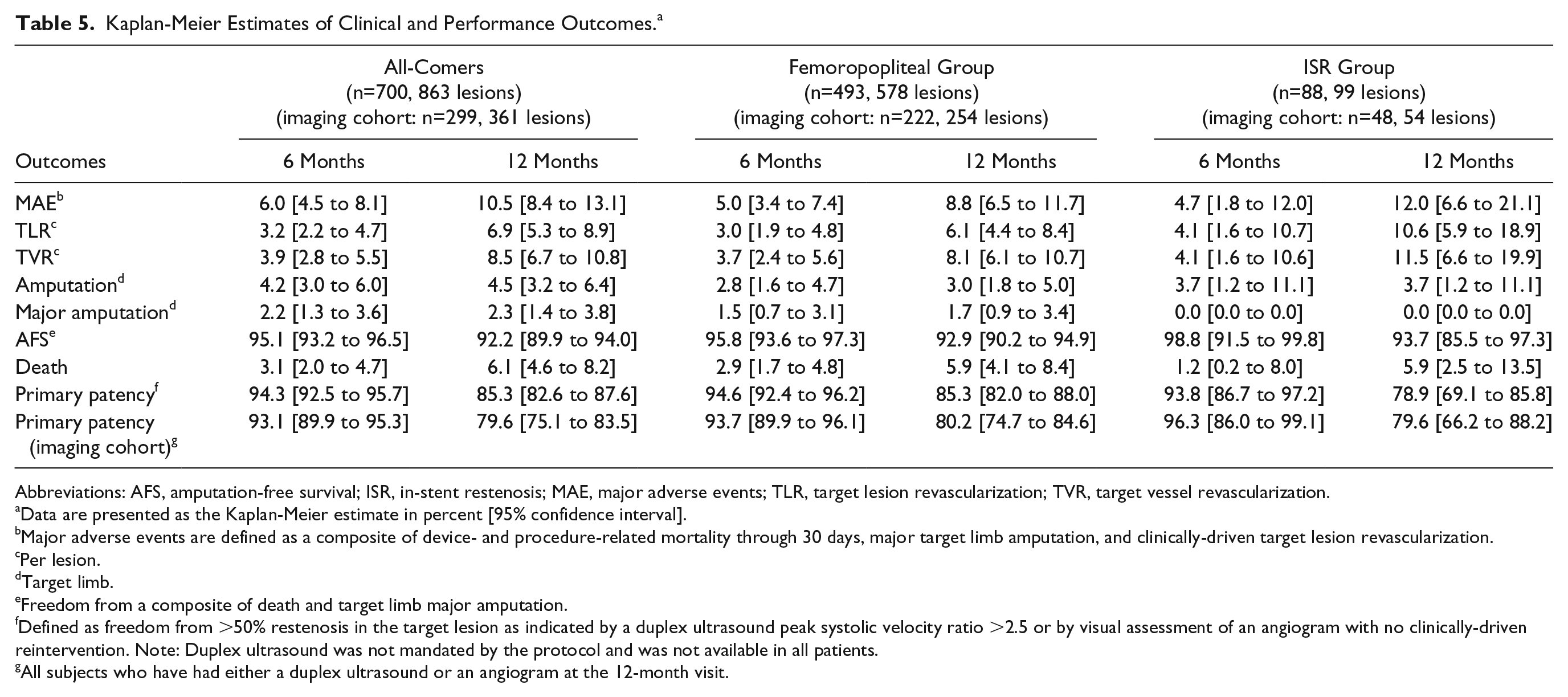
Abbreviations: AFS, amputation-free survival; ISR, in-stent restenosis; MAE, major adverse events; TLR, target lesion revascularization; TVR, target vessel revascularization.
Data are presented as the Kaplan-Meier estimate in percent [95% confidence interval].
Major adverse events are defined as a composite of device- and procedure-related mortality through 30 days, major target limb amputation, and clinically-driven target lesion revascularization.
Per lesion.
Target limb.
Freedom from a composite of death and target limb major amputation.
Defined as freedom from >50% restenosis in the target lesion as indicated by a duplex ultrasound peak systolic velocity ratio >2.5 or by visual assessment of an angiogram with no clinically-driven reintervention. Note: Duplex ultrasound was not mandated by the protocol and was not available in all patients.
All subjects who have had either a duplex ultrasound or an angiogram at the 12-month visit.
At 12 months, primary patency was 85.3% (95% CI 82.0% to 88.0%) in the femoropopliteal subgroup (Figure 4B) and 78.9% (95% CI 69.1% to 85.8%) in the ISR subset (Table 5). The patency estimates in the imaging cohorts were 80.2% (95% CI 74.7% to 84.6%) in the femoropopliteal subgroup and 79.6% (95% CI 66.2% to 88.2%) in the ISR subgroup (Table 5).
At 12 months, 8 (1.6%) subjects had a major target limb amputation in the femoropopliteal subgroup. All amputations were adjudicated by the CEC as not device related. None of the patients treated with DCB for ISR underwent a major target limb amputation through 12 months. In the femoropopliteal subgroup, 28 (5.7%) patients died within the first year, including 14 Rutherford category 5 or 6 (Figure 5B). Excluding patients with CLTI, 1-year mortality in the femoropopliteal subgroup was 3.4% (95% CI 1.9% to 6.3%). In the ISR subgroup, 5 patients had died at 12 months, corresponding to a mortality estimate of 5.9% (95% CI 2.5% to 13.5%; Table 5).
As in the all-comers cohort, ABI and the Rutherford category significantly improved at 12 months compared to baseline (Table 4).
Discussion
Compared with other DCB registries,16–19 the BIOLUX P-III all-comers registry had very few exclusion criteria, allowing the use of Passeo-18 Lux DCB in a broad population and under real-world condition. The lesions were treated per the Passeo-18 Lux instructions for use and the local standard practices at the sites, with no restriction on the use of additional devices. Thus, two-thirds of the lesions were predilated with an uncoated balloon, and about 10% of the lesions were also pretreated with other (mainly debulking) devices (rotational thrombectomy, atherectomy, scoring/cutting balloons). Although three-quarters of the lesions in the all-comers cohort and the femoropopliteal subgroup were reported as calcified by the sites, bailout stenting was required in 16.8% of the all-comers cases and 20.2% in the femoropopliteal subgroup. These procedure outcomes are within the range of comparable femoropopliteal DCB registries (17.3% to 25.3%).16–19 In the ISR subgroup, additional stents were implanted in 6.1% of the lesions, much less than in the IN.PACT ISR 20 cohort (13.4%) and DEBATE ISR 21 (15.9%). Only about half of the ISR lesions were predilated, and debulking was infrequently used to prepare the vessel prior to DCB application.
The all-comers cohort comprises a broad range of clinical presentations, including high-risk patients such as diabetics and those with CLTI. The percentage of patients having CLTI (37/7%) is, as expected, not only higher than the rates observed in pivotal randomized studies3–7 but also greater than the CLTI incidences in similar large DCB registries.16–19 This was mainly due to the exclusion of Rutherford categories 5 and 6, as well as infrapopliteal lesions in those studies. CLTI is usually the result of multilevel disease affecting the superficial femoral artery and below-the-knee popliteal artery. 22 Hence, in the femoropopliteal subgroup of the all-comers cohort, about a third of the patients had CLTI, with >20% having Rutherford category >4.
In this highly challenging patient population, the 1-year outcomes of the BIOLUX P-III registry showed that the use of the Passeo-18 Lux DCB is safe and effective. The 12-month MAE rates were low (<12%) in the all-comers cohort and subgroups, while freedom from clinically-driven TLR was 93.1% in the all-comers cohort and similar in the subgroups. Despite a higher percentage of CLTI patients and almost 12% below-the-knee lesions in BIOLUX P-III subjects, these results are consistent with other DCB registries. The 12-month freedom from clinically-driven TLR was 89.0% in the Ranger registry, 19 92.6% in the IN.PACT Global registry, 18 94.1% in the Lutonix Global registry, 17 and 94.8% in the ILLUMENATE Global registry. 16 Though in shorter lesions, freedom from clinically-driven TLR at 12 months in the ISR subgroup (89.4%) compares well with the DEBATE-ISR all-comers registry (86%) 21 and the IN.PACT Global ISR cohort (92.9%). 20 While BIOLUX P-III femoropopliteal lesions were more complex compared with BIOLUX P-I, 4 the TLR rate was reduced by more than half in BIOLUX P-III (6.1% vs 15.4%). This difference might be explained by the small patient numbers in BIOLUX P-I as well as the peak in TLR around 6 months, likely related to mandated angiography.
The major target limb amputation estimates at 12 months were 2.3% in the all-comers cohort and 1.7% in the femoropopliteal subgroup. These rates are higher than data reported from other global DCB registries,16–19 which is likely attributed to the significant percentage of CLTI and diabetic patients in BIOLUX P-III. The 1-year Kaplan-Meier mortality estimates of 6.1% in the all-comers cohort and 5.9% in the femoropopliteal subgroup are above the fatality rates reported in similar DCB registries: 0.6% in the ILLUMENATE Global study, 16 2.8% in the Lutonix Global trial, 17 and 3.5% in the IN.PACT Global study. 18 The recently published meta-analysis 23 of RCTs investigating paclitaxel-coated balloons and stents for the treatment of atherosclerotic femoropopliteal arteries also reported lower crude mortality at 12 months (2.3%). However, unlike these registries or the meta-analyzed RCTs designed to demonstrate DCB safety and efficacy in a restricted population, BIOLUX P-III allowed more severe patients to be enrolled. Hence, when limiting the BIOLUX P-III femoropopliteal subgroup to patients with intermittent claudication, which is the most comparable subgroup to the patient population included in these 3 registries and RCTs, the mortality at 1 year was 3.4%. The present data report 12-month outcomes in the all-comers cohort, while in the Katsanos meta-analysis, 23 the potential for an increased mortality risk associated with paclitaxel devices arises after the first year. The upcoming publication of BIOLUX P-III 24-month results in the full-cohort will provide insight about Passeo-18 Lux paclitaxel-coated balloon mortality risk over a greater follow-up.
Limitations
The BIOLUX P-III registry had limitations inherent to a registry, such as lack of randomization, which limits the comparability to other devices. Registries also have the possibility of underreporting. However, the selected clinical endpoints are prominent events, reducing the risk that they were overlooked by the sites. Furthermore, data from at least 25% of enrolled patients were fully monitored, and all MAEs and TLR were adjudicated by a CEC to minimize the risk of underreporting.
A major limitation was that freedom from >50% restenosis could not be systematically assessed, as by default observational registries allow only treatment according to standard of care. Therefore, the patency rate in the non-predefined imaging cohort, meaning only patients with imaging assessment performed at 12 months, is also reported. Likewise, performance outcomes such as ABI or Rutherford category assessments or questionnaires were not available for all patients. Last, the general follow-up compliance was not optimal.
Conclusion
BIOLUX P-III all-comers 12-month results confirm Passeo-18 Lux DCB safety and performance shown in the BIOLUX P-I in a large patient population with infrainguinal lesions treated under real-world condition. Despite a high-risk population, including a substantial proportion of diabetics as well as Rutherford category 5 and 6 patients, 12-month outcomes in the all-comers cohort and the femoropopliteal and ISR subgroups were good and comparable to DCB registries investigating femoropopliteal arteries. Further publication will report 24-month outcomes in the full cohort and the infrapopliteal subgroup.
Supplemental Material
19-0467_suppl_tables – Supplemental material for Paclitaxel-Coated Balloon for the Treatment of Infrainguinal Disease: 12-Month Outcomes in the All-Comers Cohort of BIOLUX P-III Global Registry
Supplemental material, 19-0467_suppl_tables for Paclitaxel-Coated Balloon for the Treatment of Infrainguinal Disease: 12-Month Outcomes in the All-Comers Cohort of BIOLUX P-III Global Registry by Gunnar Tepe, Thomas Zeller, Matej Moscovic, Jean-Marc Corpataux, Johnny Kent Christensen, Koen Keirse, Giovanni Nano, Henrik Schroeder, Christoph A. Binkert and Marianne Brodmann in Journal of Endovascular Therapy
Footnotes
Acknowledgements
The authors would like to acknowledge the important contribution of all BIOLUX P-III centers and study personnel and the support of BIOTRONIK in organizing the registry and preparing the manuscript.
Authors’ Note
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Gunnar Tepe has received study support from B. Braun, Bard, Bayer, Biotronik, Boston Scientific Corp., W. L. Gore & Associates, Medtronic, Philips, Verian, and Shockwave Medical Inc. Thomas Zeller has received honoraria from Abbott Vascular, Veryan, Biotronik, Boston Scientific Corp., Cook Medical, W. L. Gore & Associates, Medtronic, Philips-Spectranetics, TriReme, and Shockwave; he is a consultant for Boston Scientific Corp., Cook Medical, W. L. Gore & Associates, Medtronic, Spectranetics, Veryan, Intact Vascular, B. Braun, Shockwave, Bayer, and Vesper Medical; his institution has received research, clinical trial, or drug study funds from 480 Biomedical, Bard Peripheral Vascular, Veryan, Biotronik, Cook Medical, W. L. Gore & Associates, Medtronic, Philips, Terumo, TriReme, Shockwave, Med Alliance, Intact Vascular, and B. Braun; and he owns common stock in QT Medical. Christoph Binkert is a consultant to Merit Medical and receives research support from Philips/Profound and Abbott. Marianne Brodmann has received honoraria from Abbott Vascular, Biotronik, Philips-Spectranetics, Medtronic, Daiichi Sankyo, Bayer Healthcare, and BD Bard; she is a consultant to Boston Scientific Corp., Medtronic, Spectranetics, Intact Vascular, Shockwave, Bayer, Vesper Medical, and BD Bard; and her institution has received research, clinical trial, or drug study funds from 480 Biomedical, BD Bard, Biotronik, Medtronic, Philips, Shockwave, Med Alliance, Intact Vascular, and B. Braun. All authors are clinical investigators in the BIOLUX P-III registry.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This registry was funded by BIOTRONIK AG, Buelach, Switzerland.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.