
Abstract
Aims:
Nephron-sparing surgery (NSS) is associated with acute kidney injury (AKI) that can accelerate chronic kidney disease progression, yet the mechanisms driving NSS-related injury (NRI) remain incompletely understood. In particular, how surgical stress reprograms proximal tubular energy metabolism independent of persistent ischemia remains unresolved. This study aimed to elucidate the mechanisms underlying lipid metabolic dysregulation and mitochondrial dysfunction in NRI.
Results:
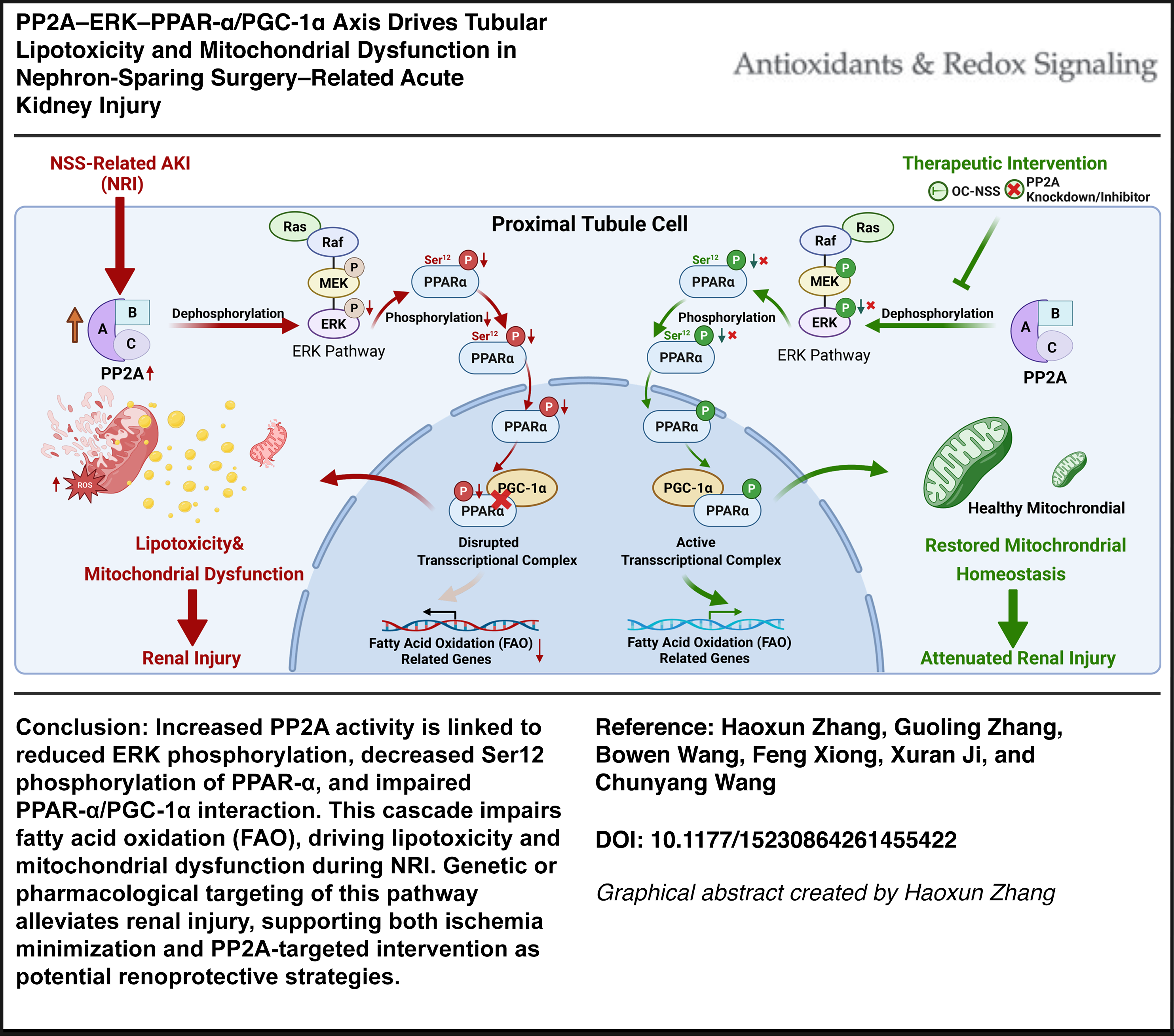
Clinical cohort analysis showed that prolonged warm ischemia time was independently associated with postoperative AKI. Across human and experimental NRI, proximal tubular cells exhibited consistent suppression of fatty acid oxidation, increased lipid accumulation, and mitochondrial dysfunction. These metabolic abnormalities were associated with increased protein phosphatase 2A (PP2A) activity, reduced extracellular signal-regulated kinase (ERK) phosphorylation, and diminished transcriptional activity of the peroxisome proliferator-activated receptor-alpha (PPAR–α)/PGC-1α metabolic axis. Experimental modulation of PP2A or restoration of ERK-dependent PPAR-α signaling partially rescued mitochondrial function, reduced lipotoxic stress, and attenuated tubular injury. Single-cell and cell-type-resolved analyses localized these changes predominantly to metabolically active proximal tubular subpopulations, supporting the cell-type specificity of this pathway.
Innovation and Conclusion:
This study identifies PP2A-dependent suppression of ERK–PPAR-α/PGC-1α signaling as a central mechanism underlying proximal tubular metabolic failure in NRI. By linking surgical stress to disordered lipid metabolism, these findings reveal a previously underappreciated regulatory pathway distinct from classical ischemic injury and highlight tubular metabolic resilience as a therapeutic target for mitigating kidney injury following nephron-sparing surgery. Antioxid. Redox Signal. 00, 000–000.
Keywords
Get full access to this article
View all access options for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.