
Abstract
Aims:
Chronic inflammation is a widely acknowledged contributor to the development of atherosclerosis. Gasdermin D (GSDMD) serves as a key executor of pyroptosis in inflammatory diseases. This study aims to determine the role of endothelial GSDMD in lipopolysaccharide (LPS)-accelerated atherosclerosis and elucidate its underlying molecular mechanisms.
Results:
GSDMD expression was aberrantly activated in both LPS-accelerated atherosclerotic animal models and oxidized low-density lipoprotein plus LPS-treated endothelial cell models. Compared with the control, endothelial GSDMD deficiency attenuated the atherogenesis progression and vascular endothelial inflammation induced by LPS and protected against the progression of mitochondrial damage, the release of mitochondrial ROS and mitochondrial DNA, and the activation of the stimulator of interferon genes (STING) pathway both in vivo and in vitro. Mechanistically, endothelial GSDMD expression mediates mitochondrial membrane permeabilization and mitochondrial damage-associated molecular patterns release and triggers the STING pathway to aggravate atherosclerotic progression. In addition, the STING pathway activation was proved to partially reverse the effects of endothelial GSDMD deficiency both in vivo and in vitro. Moreover, the signal transducer and activator of transcription 3 was identified as a positive regulator of GSDMD expression.
Innovation and Conclusion:
Our findings elucidate the mechanism by which endothelial GSDMD exerts its atherogenic effects by increasing mitochondrial damage and upregulating the STING pathway in LPS-accelerated atherosclerosis. GSDMD promises to be a critical therapeutic target for atherosclerotic cardiovascular diseases. Antioxid. Redox Signal. 44, 61–84.
This is a visual representation of the abstract.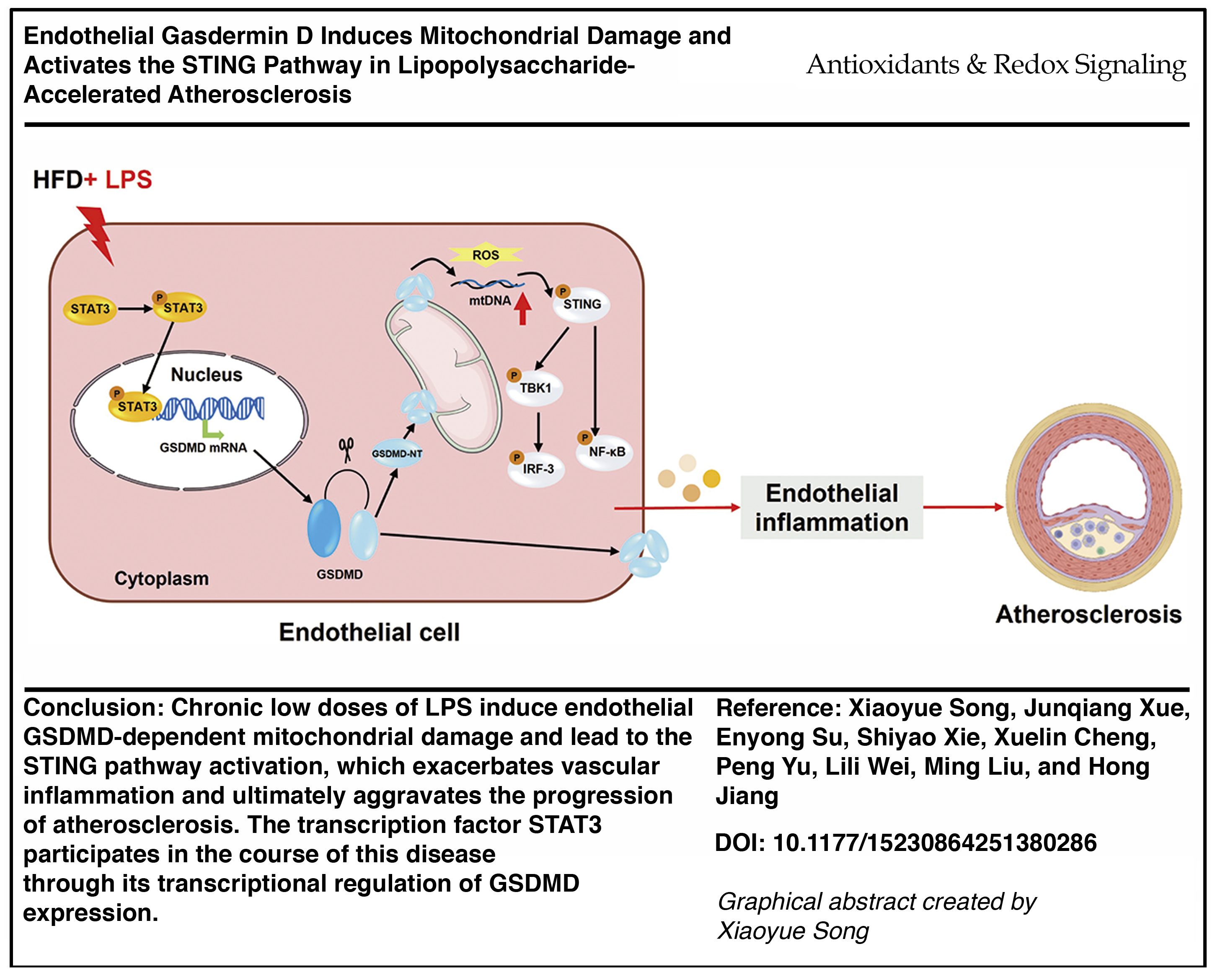
Introduction
Chronic inflammation characterized by abnormal lipid metabolism and atherosclerotic plaque formation in the intimal spaces of vessels is the hallmark of atherosclerosis (Libby and Hansson, 2019). Despite substantial advancements in clinical and experimental research, atherosclerotic diseases continue to rank as the global leading causes of death (Roth et al., 2017). In addition to conventional risk factors, chronic infection has been closely linked to the worsening of atherosclerosis (Shah, 2019). For instance, an association between chronic low-grade inflammatory conditions such as periodontitis and atherosclerosis has been reported (Sanz et al., 2020). Experimental animal data have confirmed that long-term lipopolysaccharide (LPS) infusions increase atherosclerotic plaque areas and aggravate the inflammatory infiltration in plaques (Lehr et al., 2001; Ostos et al., 2002). Research on subclinical endotoxemia has revealed that low-density lipoprotein (oxidized low-density lipoprotein [ox-LDL]) and LPS cooperatively activate macrophages to express pro-inflammatory cytokines (Wiesner et al., 2010). Previous studies have indicated that the endothelial cytotoxicity of LPS contributes to vascular inflammation (Cheng et al., 2017). Mechanistically, endothelial cell injury is regarded as an initial critical step in the pathogenesis of atherosclerosis (Xu et al., 2021). The mechanisms underlying the potential impacts of ox-LDL and low doses of LPS on endothelial cells remain incompletely understood. To reveal the synergistic effects in the context of atherosclerosis and chronic inflammatory conditions, we must mimic the long-term low-grade inflammatory effects of ox-LDL and low doses of LPS on endothelial cells.
Conclusions and Innovation
In summary, our findings reveal that GSDMD is a prominent protein activated in chronic low-dose LPS-accelerated atherosclerosis and holds promise as a potentially effective target for the prevention and treatment of atherosclerotic diseases combined with chronic inflammation. Mechanistically, we showed that the activation of endothelial GSDMD triggered the cGAS-STING pathway activation through mitochondrial membrane permeabilization and mtROS and mtDNA release into the cytosol, ultimately aggravating the progression of atherosclerosis.
Gasdermin D (GSDMD), belonging to the gasdermin protein family, serves as a key executor of pyroptosis in inflammatory diseases. Upon activation, cysteinyl aspartate specific proteinase (Caspase)-4/5/11 cleaves GSDMD to generate the N-terminal (GSDMD-NT) and C-terminal domains (Shi et al., 2015). GSDMD-NT oligomerizes in the plasma membrane to form pyroptosis-related pores and promotes the secretion of the mature forms of interleukin (IL)-1β and IL-18 (Burdette et al., 2021). Several studies have demonstrated that pyroptosis is involved in the initiation and progression of atherosclerosis (Qian et al., 2021). In our previous work, we showed that pharmacological suppression of GSDMD attenuated vascular inflammation and atherosclerotic lesions in apolipoprotein E (ApoE)-deficient mice (Zhang et al., 2023a). In view of the crucial role of GSDMD in atherosclerosis and sepsis, further investigations on the mechanism of GSDMD-mediated endothelial cell injury in LPS-induced atherosclerosis are warranted.
Mitochondria are the principal sites of oxidative metabolism and energy metabolism (Liu et al., 2014). Previous observations have implied an association between mitochondrial damage and the progression of atherosclerotic lesions (Salnikova et al., 2021). For instance, mitochondrial DNA (mtDNA) damage within atherosclerotic plaques from animal models and clinical samples leads to reduced mitochondrial respiration, resulting in increased necrotic core formation and reduced fibrous cap thickness (Khwaja et al., 2021). The cyclic guanosine monophosphate–adenosine monophosphate synthase stimulator of interferon genes (cGAS-STING) signaling axis was originally shown to trigger an innate immune reaction following its detection of pathogenic DNA (Ishikawa and Barber, 2008). Upon cGAS-STING activation, STING recruits TANK-binding kinase 1 (TBK1) and phosphorylates STING and interferon regulatory factor 3 (IRF-3) (Decout et al., 2021). STING activation also leads to the activation of nuclear factor kappa-B (NF-κB), triggering the production of pro-inflammatory cytokines (Abe and Barber, 2014). There is evidence that STING expression plays a pivotal role in atherogenesis in response to endogenously released mtDNA fragments (Pham et al., 2021; Ueda et al., 2023). Interestingly, GSDMD-NT can also be located in the mitochondrial membrane, triggering mitochondrial damage, releasing mtDNA, and activating the cGAS-STING pathway (Huang et al., 2020).
In the present study, we first examined the aberrant activation of GSDMD expression in chronic low-dose LPS-accelerated atherosclerosis and reported its colocalization with the endothelium. GSDMD deficiency was found to suppress vascular inflammation and atherosclerotic lesions. The protective role of GSDMD inhibition was also confirmed in endothelial injury models. Then we explored the mechanism of mitochondrial damage mediated by GSDMD. Next, we investigated the upregulation of the cGAS-STING pathway in in vivo and in vitro models of chronic low-dose LPS-accelerated atherosclerosis. Furthermore, we performed in vivo and in vitro rescue experiments to explore the regulatory mechanisms of downstream signaling pathways by utilizing the specific STING agonist SR-717. Moreover, a classical transcription factor, signal transducer and activator of transcription 3 (STAT3), was predicted to bind to the GSDMD promoter. In summary, our findings provide a rational explanation for the aggravation of atherogenic processes induced by chronic exposure to low doses of LPS and highlight the potential value of targeting endothelial GSDMD for the prevention and treatment of atherosclerosis.
Results
Elevated endothelial GSDMD expression in LPS-accelerated atherosclerosis
In our study, high-fat diet-fed ApoEKO mice received weekly injections of low doses of LPS from 6 to 18 weeks of age. Aorta tissues were harvested at week 6, week 10, week 14, and week 18. The results of hematoxylin-eosin (HE) staining collectively demonstrated that the area of atherosclerotic lesions increased with the increasing time following LPS injection (Fig. 1A, B). Furthermore, the extent of F4/80-positive macrophage infiltration within atherosclerotic plaques exhibited similar trends (Supplementary Fig. S1A, B). Western blot analysis revealed increased expressions of intercellular cell adhesion molecule 1 (ICAM1), vascular cell adhesion molecule 1 (VCAM1), and monocyte chemotactic protein-1 (MCP-1) (Supplementary Fig. S1C, D). These findings suggest that persistent low-grade LPS exposure contributes to the development of atherosclerosis and vascular inflammation. The plasma lipid levels of the mice increased over time, and the lipid levels of mice aged 18 weeks were significantly greater than those of 6-week-old mice. No significant difference in plasma lipid levels was observed between the groups at the same time points (Supplementary Fig. S2A). To evaluate the role of GSDMD in the development of LPS-accelerated atherosclerosis, we examined GSDMD expression in animal models. Western blot analysis revealed progressive increases in the expression and cleavage of GSDMD over time (Fig. 1C, D). Immunofluorescence staining revealed that GSDMD can colocalize with the endothelial cell marker cluster of differentiation 31 (CD31) (Fig. 1E). We found that compared with no LPS stimulation, LPS treatment accelerated atherosclerosis, leading to greater expression levels of inflammatory Caspase-1 and Caspase-11 in aortas derived from 18-week-old ApoEKO mice. The trends of the variations in Caspase-1/11 expressions were consistent with the expression of GSDMD-NT (Supplementary Fig. S3A-C).
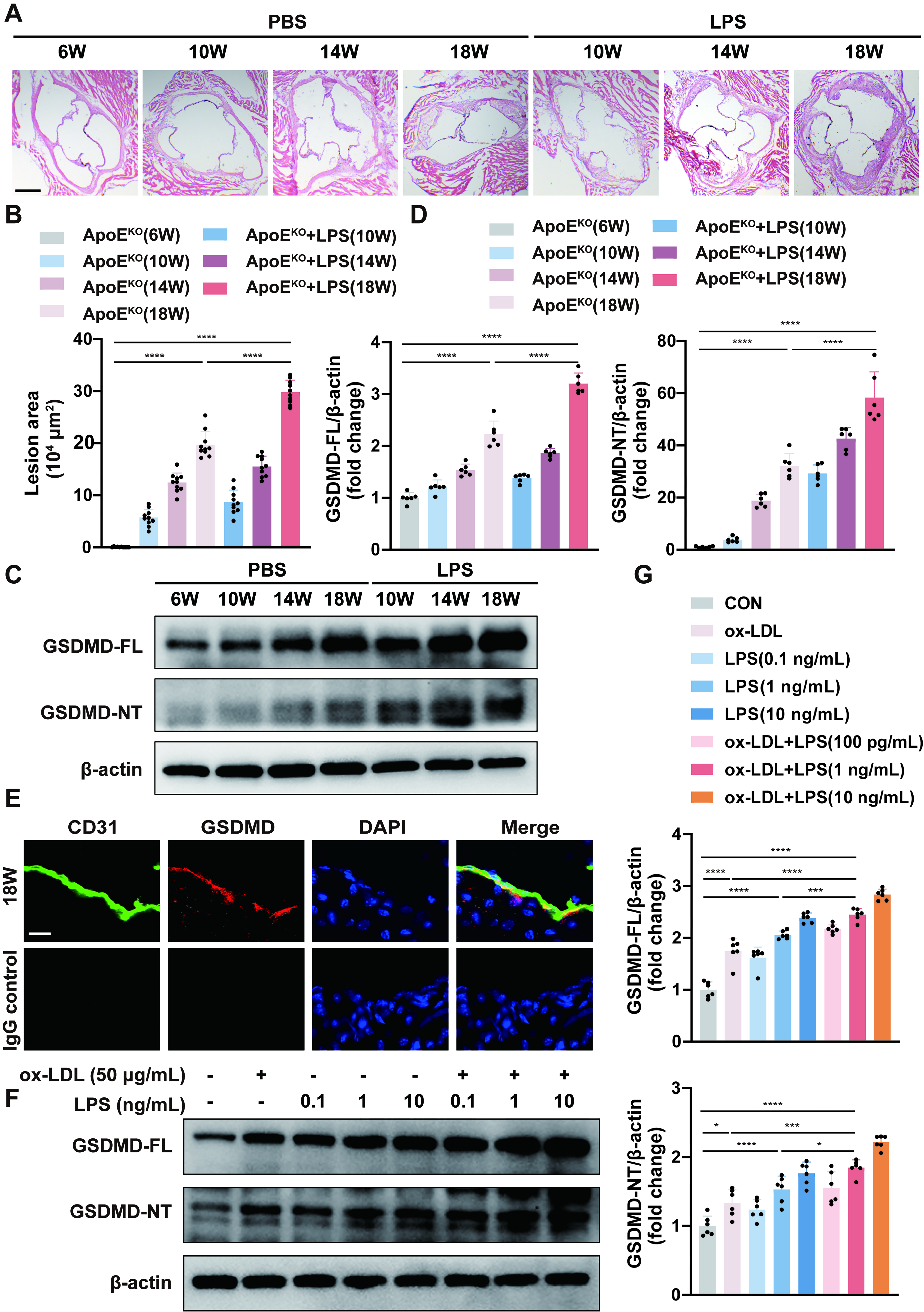
To mimic long-term low-grade inflammation in vitro, human umbilical vein endothelial cells (HUVECs) were stimulated with a combination of low doses of ox-LDL and LPS for 24 h. Different concentrations (0.1 ng/mL, 1 ng/mL, and 10 ng/mL) of LPS were chosen to determine the optimal concentration. We found that 1 ng/mL LPS was sufficient enough to induce inflammation in the endothelium and that the combination of ox-LDL and LPS significantly worsened inflammatory manifestations. We performed Western blot to observe the upregulated expressions of the adhesion molecules ICAM1 and VCAM1 (Supplementary Fig. S4A, B). A human Tohoku Hospital Pediatrics-1 (THP-1) monocyte adhesion assay also revealed increased adhesion of endothelial cells (Supplementary Fig. S4C, D). Consistent with the findings of the animal experiments, we also detected the aberrant activation of GSDMD in the cell models (Fig. 1F, G). Thus, the administration of LPS (1 ng/mL) plus ox-LDL (50 μg/mL) was used to induce endothelial injury during the progression of LPS-accelerated atherosclerosis in vitro. These results suggest that endothelial GSDMD might be involved in LPS-accelerated atherosclerosis.
GSDMD deficiency ameliorates LPS-accelerated atherogenesis progression and vascular endothelial inflammation in vivo and in vitro
Given the increased endothelial GSDMD expression in LPS-accelerated atherosclerosis, we next assessed the effects of GSDMD on atherogenesis progression and vascular inflammation. To do this, GSDMDKO/ApoEKO mice and GSDMDECKO/ApoEKO mice were generated and exposed to weekly injections of low doses of LPS from 6 weeks of age to 18 weeks of age to construct the animal models. ApoEKO mice and GSDMDWT/ApoEKO mice were used as controls. Mouse genotyping was conducted to confirm the success of the transgenic mouse construction (Supplementary Fig. S5A, B). There was no significant difference in the lipid levels of the mice among the groups (Supplementary Fig. S2B, C).
For the LPS-accelerated GSDMDKO/ApoEKO mouse models, in situ images of the aortic arches and HE staining of the aortic roots depicted decreased atherosclerotic lesion areas compared with those in the LPS-accelerated ApoEKO control mice (Supplementary Fig. S6A-C). Similarly, the number of F4/80-positive macrophages within aortic atherosclerotic lesions in LPS-accelerated GSDMDKO/ApoEKO mice was reduced (Supplementary Fig. S6D, E). We detected a significant decrease in the expressions of ICAM1, VCAM1, and MCP-1 in the aortas of LPS-accelerated GSDMDKO/ApoEKO mice by Western blot analysis (Supplementary Fig. S6F, G). Consistently, mRNA expression levels of pro-inflammatory genes such as IL-1β, IL-6, IL-18, and MCP-1 were markedly decreased in the aortas of LPS-accelerated GSDMDKO/ApoEKO mice (Supplementary Fig. S6H). In addition, the serum levels of characteristic pyroptotic inflammatory factors IL-1β and IL-18 were significantly decreased (Supplementary Fig. S7A).
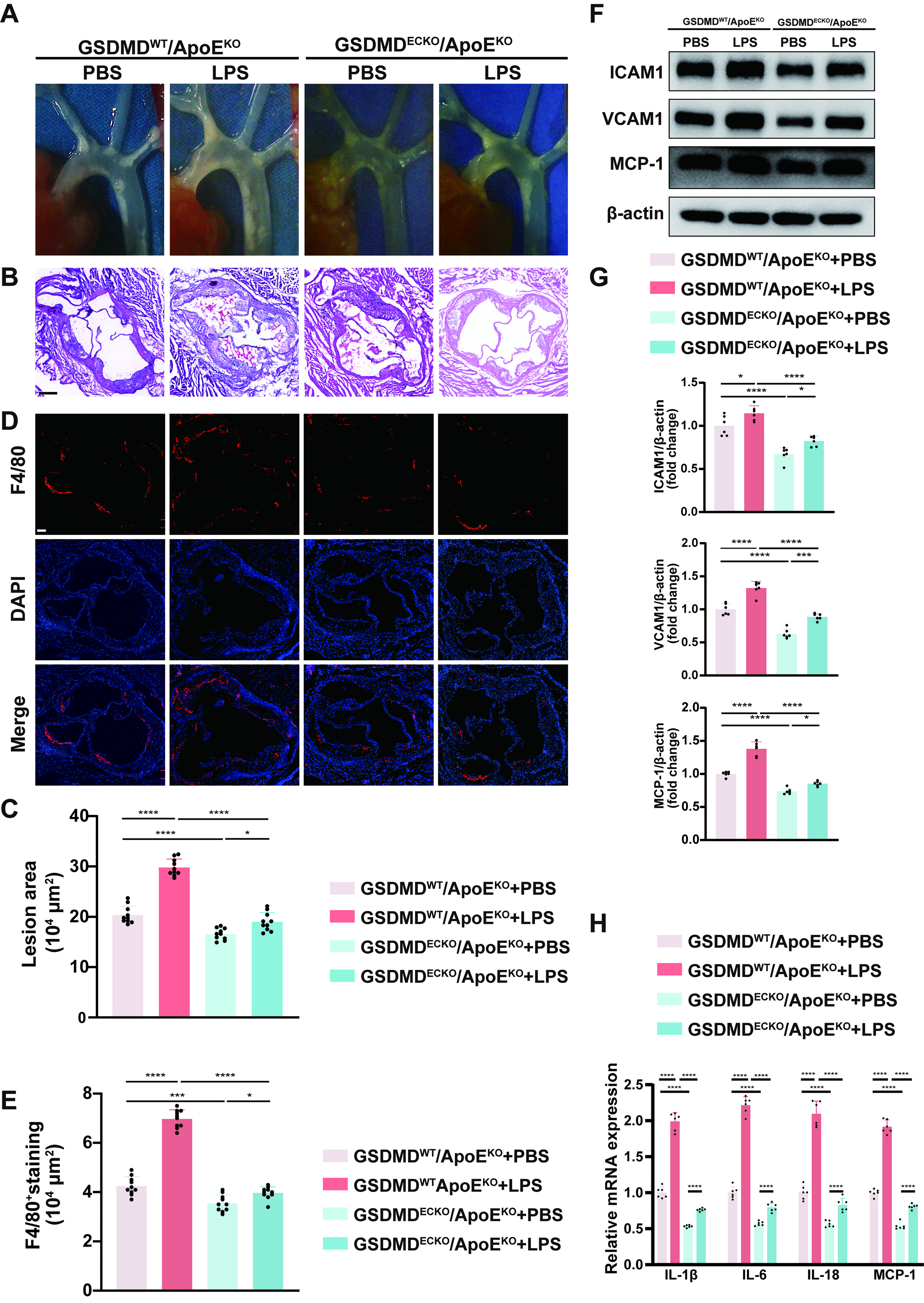
For the LPS-accelerated endothelial-specific GSDMD knockout ApoEKO mice, representative in vivo images of the aortic arches and histological analysis represented that the loss of endothelial GSDMD expression reduced atherosclerotic plaque sizes in comparison with those in LPS-accelerated GSDMDWT/ApoEKO mice (Fig. 2A–C). These mice also showed less F4/80-positive macrophage deposition than control mice, as shown by immunofluorescence staining analysis (Fig. 2D, E). In addition, the expression levels of ICAM1, VCAM1, and MCP-1 were suppressed in the aortas of LPS-accelerated GSDMDECKO/ApoEKO mice than in those of the control mice (Fig. 2F, G). Furthermore, the inhibition of endothelial GSDMD reduced the IL-1β, IL-6, IL-18, and MCP-1 mRNA levels and serum IL-1β and IL-18 levels compared with those in the control mice (Fig. 2H and Fig. S7B). These findings strongly suggest that GSDMD deficiency attenuates the atherogenesis progression and endothelial inflammation induced by LPS in vivo.
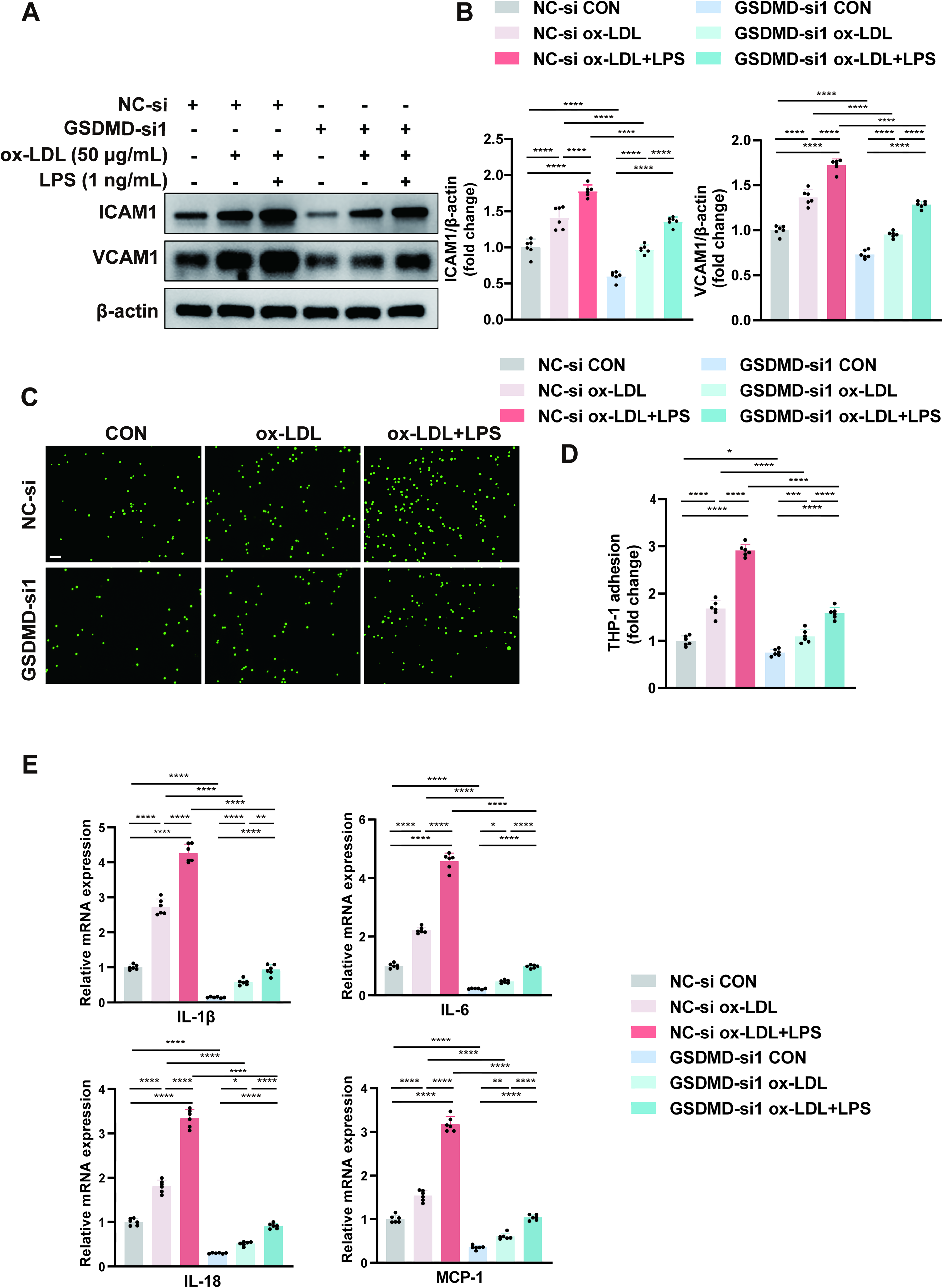
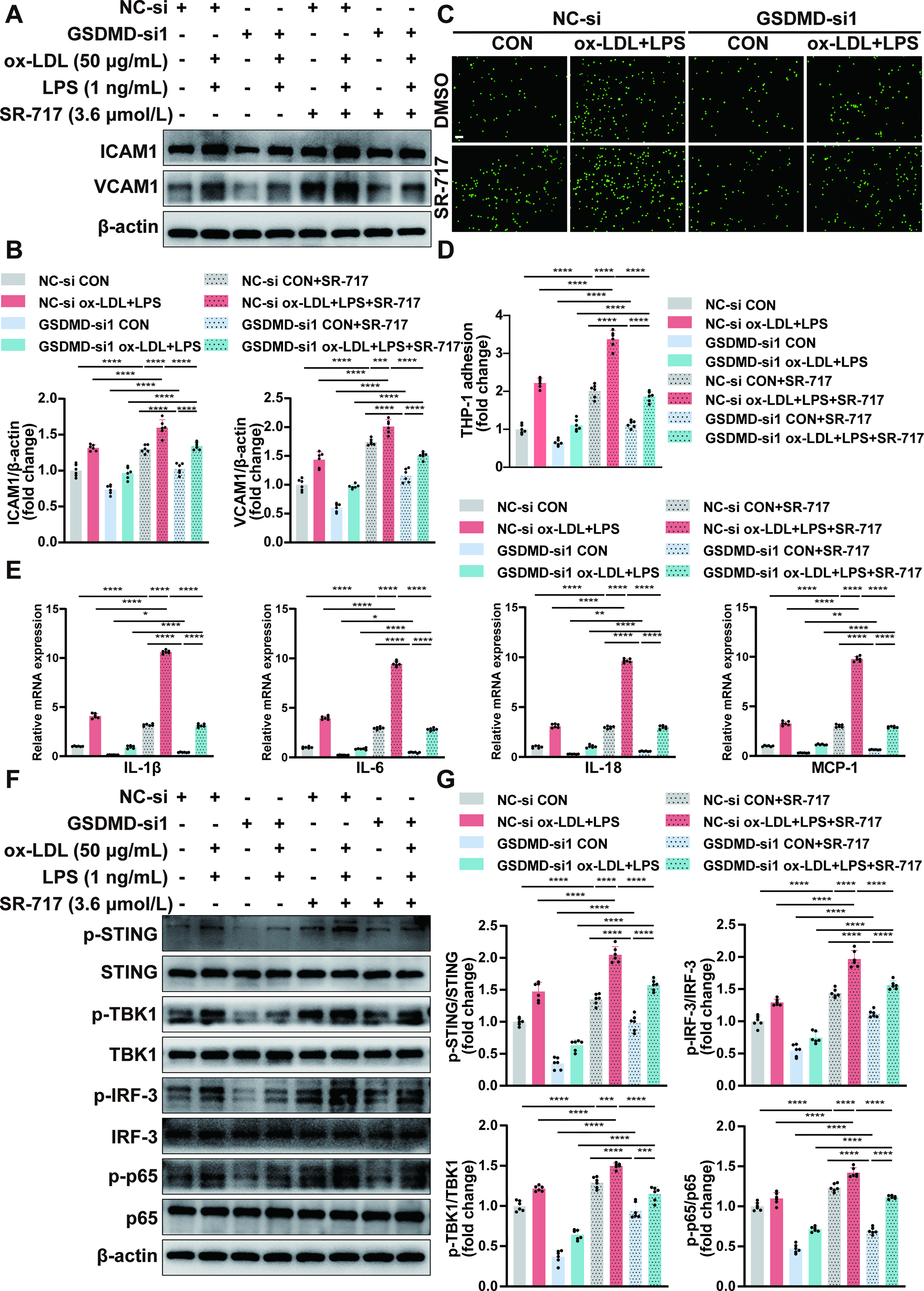
To explore the cellular mechanisms within the lesions, we constructed GSDMD knockdown HUVECs, and the silencing efficacy was confirmed by Western blot (Supplementary Fig. S8A, B). In GSDMD-knockdown HUVECs, the expression levels of ICAM1 and VCAM1 were downregulated. Among the three intervention conditions, namely, control, ox-LDL, and ox-LDL plus LPS, ox-LDL combined with LPS had the greatest effect on the protein expression levels, followed by ox-LDL alone and the control (Fig. 3A, B). We observed significantly reduced adherence of Calcein AM–labeled THP-1 cells to GSDMD-knockdown HUVECs under the three intervention conditions described above (Fig. 3C, D). Real-time quantitative polymerase chain reaction (qPCR) analysis revealed similar trends in the mRNA expression levels of IL-1β, IL-6, IL-18, and MCP-1 compared with those in the GSDMD-knockdown HUVECs subjected to the different interventions described above (Fig. 3E). We also observed a significant reduction in IL-1β and IL-18 secretion in the supernatants from the GSDMD-knockdown HUVECs by enzyme-linked immunosorbent assay (ELISA) and lactate dehydrogenase (LDH) release levels by LDH release assay (Supplementary Fig. S9A, C). Our data strongly suggest that GSDMD deficiency attenuates the atherogenesis progression and endothelial inflammation induced by LPS stimulation in vitro. Altogether, these results identify endothelial GSDMD as an underlying mediator of atherogenesis progression and endothelial inflammation in LPS-accelerated atherosclerosis.
GSDMD ablation protects against mitochondrial damage in vivo and in vitro
Numerous mitochondrial components and metabolites can be released as damage-associated molecular patterns and promote inflammatory responses (Marchi et al., 2023). Immunofluorescence staining of aortic tissues for cytosolic double-stranded DNA (dsDNA) and the endothelial cell marker CD31 revealed increased dsDNA release in endothelial cells within LPS-accelerated atherosclerotic lesions. GSDMD ablation protected against dsDNA release into the cytosol in LPS-accelerated GSDMDKO/ApoEKO mice and LPS-accelerated GSDMDECKO/ApoEKO mice (Fig. 4A, B). To verify whether the atherogenic effects of low-grade persistent LPS exposure are associated with mitochondrial damage, we performed a series of in vitro experiments to assess the mitochondrial membrane potential (MMP), mitochondrial permeability transition pore (MPTP) opening, and mitochondrial reactive oxygen species (mtROS) contents. Tetramethylrhodamine ethyl ester (TMRE) staining revealed that the red fluorescence intensity significantly decreased after treatment with ox-LDL and LPS, which indicated a decrease in the MMP. GSDMD ablation partially rescued the MMP (Fig. 4C). Moreover, the shift in JC-1 staining from red to green fluorescence was also an indicator of a decreased MMP. An increase in green fluorescence was accompanied by a decrease in red fluorescence after stimulation with ox-LDL and LPS. GSDMD ablation reversed the decrease in the MMP caused by the abovementioned stimulation (Fig. 4D). With respect to MPTP opening, green fluorescence was significantly decreased after exposure to ox-LDL and LPS, which aggravated MPTP opening, and this effect was mitigated by GSDMD ablation (Fig. 4E). MitoSOX staining showed that the red fluorescence intensity was overtly increased after stimulation with ox-LDL and LPS, which represented increased mtROS production, and this effect was reversed by GSDMD ablation (Fig. 4F). These findings suggest that GSDMD expression is related to the mitochondrial damage caused by LPS-accelerated atherosclerosis.
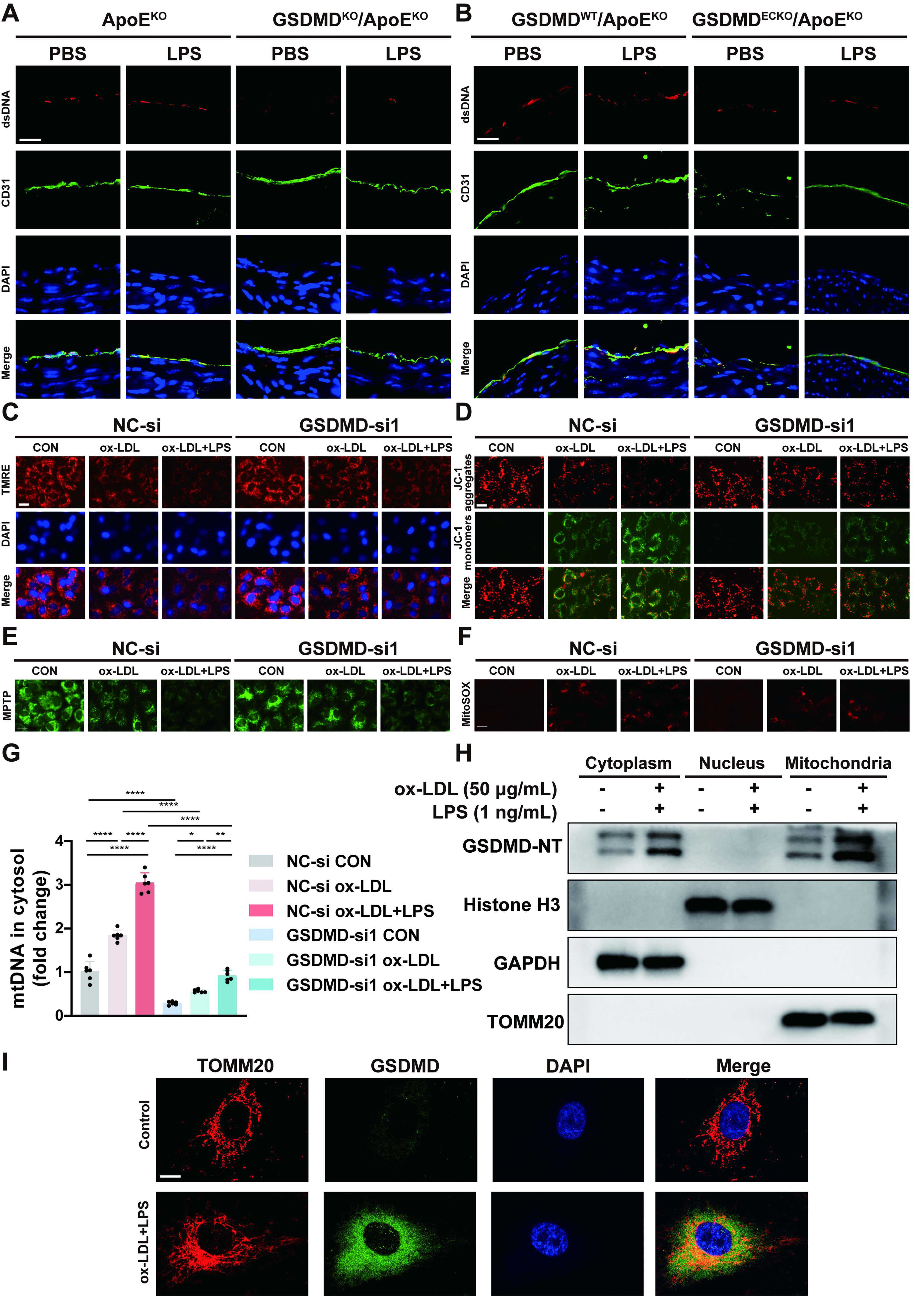
Because cytosolic dsDNA can be either mtDNA released from damaged mitochondria or nuclear DNA (nDNA) released from damaged nucleus, qPCR analysis was used to determine the ratio of nicotinamide adenine dinucleotide dehydrogenase subunit 1 (ND1) expression (a mtDNA marker) to β-globin expression (an nDNA marker) in the cytosolic fraction. We observed an increase in the cytoplasmic mtDNA/nDNA ratio in ox-LDL plus LPS-treated HUVECs, whereas GSDMD-small interfering RNA (siRNA) suppressed mtDNA leakage into the cytosol (Fig. 4G). Taken together, these data indicate that the dsDNA released into the cytosol is derived primarily from damaged mitochondria.
Several studies have demonstrated that GSDMD-NT can be enriched in the mitochondrial membrane and facilitate pore formation and mtDNA leakage, resulting in mitochondrial damage. Western blot analysis revealed that GSDMD-NT was localized to the mitochondrial fraction after stimulation with ox-LDL and LPS (Fig. 4H). Immunofluorescence staining of GSDMD and translocase of outer mitochondrial membrane 20 homolog (TOMM20) in ox-LDL plus LPS-treated HUVECs also exhibited a colocalization relationship (Fig. 4I). Thus, the characteristics of mitochondrial localization and mitochondrial pore formation of GSDMD-NT are indispensable for dsDNA release into the cytosol. These results collectively suggest that endothelial GSDMD expression mediates mitochondrial damage and mtROS and mtDNA release in LPS-accelerated atherosclerosis.
GSDMD inhibition attenuates STING pathway activation in vivo and in vitro
Recent studies have shown that cytoplasmic DNA, a universal damage signal, can be recognized by cGAS sensors, resulting in the synthesis of cyclic guanosine monophosphate–adenosine monophosphate (cGAMP) and subsequent activation of the STING pathway (Chen and Xu, 2023). Western blot results indicated that the phosphorylation levels of STING, TBK1, IRF-3, and p65 were substantially elevated in LPS-accelerated atherosclerosis, whereas no differences were observed in the total protein levels of STING, TBK1, IRF-3, and p65. Hence, the expression levels of p-STING/STING, p-TBK1/TBK1, phospho-IRF-3 (p-IRF-3)/IRF-3, and p-p65/p65 were significantly augmented in vivo under these stimuli. In the LPS-accelerated GSDMDKO/ApoEKO mouse models, we detected an obvious decrease in expression levels of p-STING, p-TBK1, p-IRF-3, and p-p65 in aortas via Western blot analysis compared with those in the LPS-accelerated ApoEKO control mice. However, the total protein expressions of STING, TBK1, IRF-3, and p65 remained unchanged, leading to a reduced ratio of phosphorylated proteins to total proteins (Fig. 5A, B). In LPS-accelerated endothelial-specific GSDMD knockout ApoEKO mice, the expression levels of p-STING, p-TBK1, p-IRF-3, and p-p65 were lower than those in LPS-accelerated GSDMDWT/ApoEKO control mice (Fig. 5C, D). These findings strongly suggest that GSDMD inhibition attenuates the activation of the STING pathway in vivo.
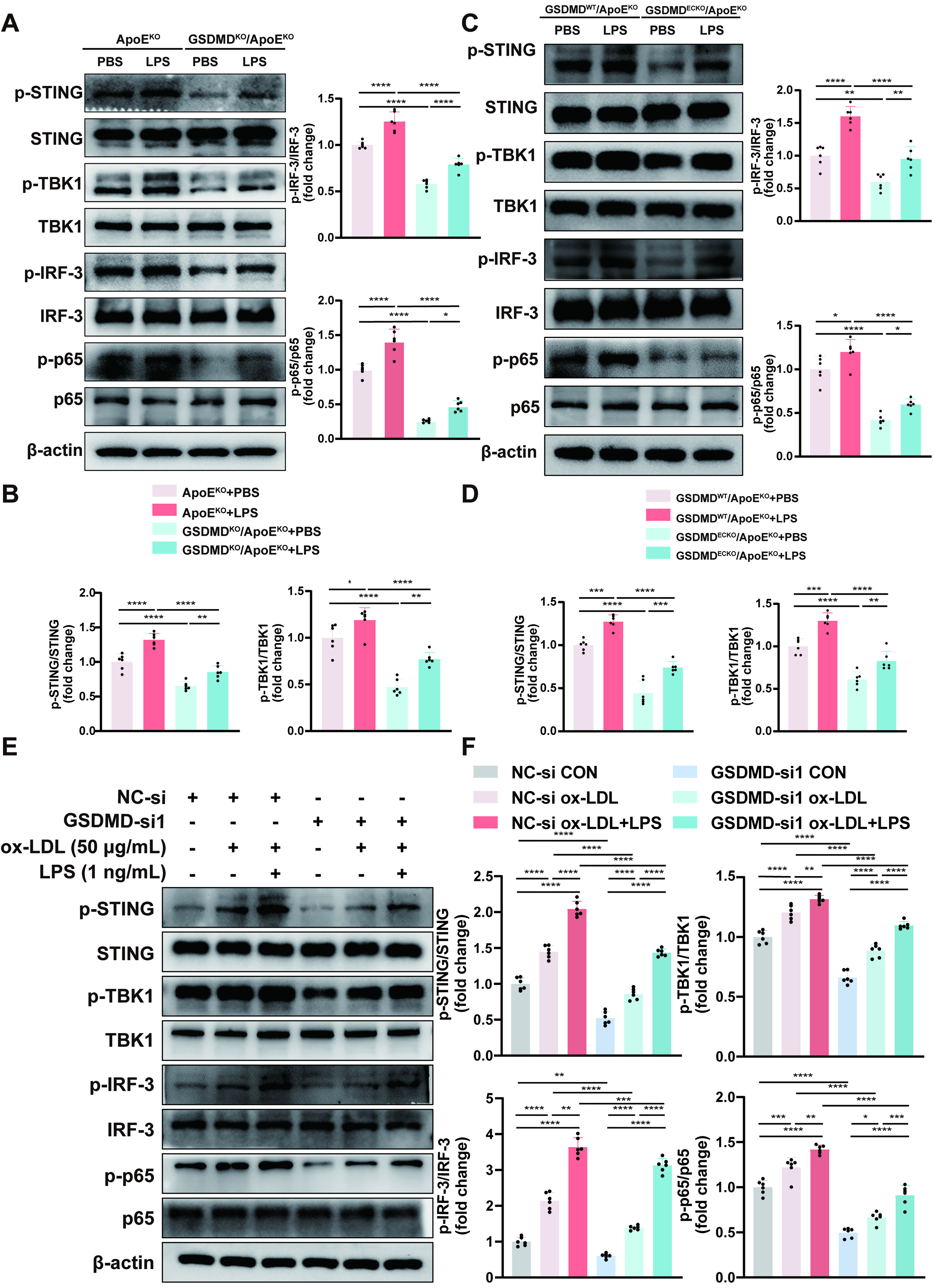
We then investigated the activation of the STING pathway in HUVECs incubated under different intervention conditions and found that the expressions of p-STING, p-TBK1, p-IRF-3, and p-p65 were upregulated, whereas the expressions of STING, TBK1, IRF-3, and p65 were unchanged. Among the three intervention conditions, namely, the control, ox-LDL, and ox-LDL plus LPS groups, the phosphorylation levels of STING, TBK1, IRF-3, and p65 were the highest with the combination of ox-LDL and LPS, followed by the ox-LDL and control groups. Thus, the expression levels of p-STING/STING, p-TBK1/TBK1, p-IRF3/IRF3, and p-p65/p65 increased after incubation under the three intervention conditions described above. In the GSDMD-knockdown HUVECs, the elevated expressions of p-STING, p-TBK1, p-IRF-3, and p-p65 decreased under the three intervention conditions described above, with no effect on the total protein expression levels (Fig. 5E, F). Our data strongly suggest that GSDMD inhibition attenuates STING pathway activation in vitro. Taken together, these findings strongly revealed that endothelial GSDMD regulates the activation of the STING pathway in LPS-accelerated atherosclerosis.
GSDMD blockade can be reversed by the activation of the STING pathway in vivo and in vitro
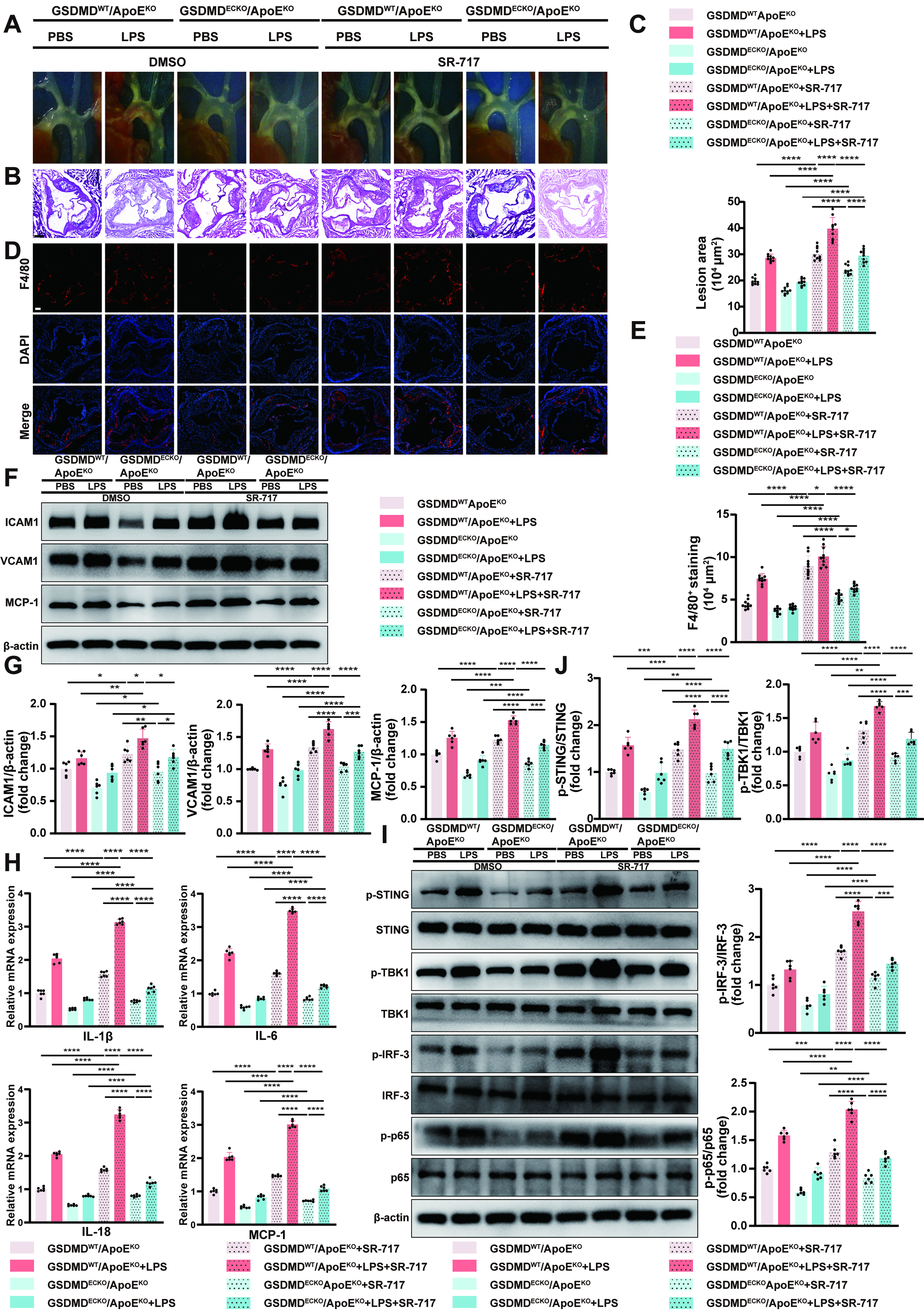
To elucidate the molecular mechanisms by which endothelial GSDMD regulates the STING pathway, we used a specific STING agonist (SR-717) to upregulate the activity of the STING pathway in our experiments, especially to observe the effects of the agonist on the GSDMD-knockdown background.
With respect to LPS-accelerated endothelial-specific GSDMD knockout ApoEKO mice, in situ images of the aortic arches and HE staining of the aortic roots depicted that SR-717 administration led to the extension of atherosclerotic lesions (Fig. 6A–C). These findings suggest that the activation of the STING pathway by SR-717 can partially reverse the inhibitory effects on atherogenesis caused by GSDMD blockade. Lipid levels did not significantly differ among the groups (Supplementary Fig. S2D). Immunofluorescence staining of F4/80-positive macrophages revealed an increase in the number of target cells within the atherosclerotic plaques after daily injections of SR-717 for four weeks (Fig. 6D, E). Western blot analysis revealed that the expressions of ICAM1, VCAM1, and MCP-1 increased in the aortas (Fig. 6F, G). Consistently, the increases in the IL-1β, IL-6, IL-18, and MCP-1 mRNA levels and serum IL-1β and IL-18 levels were determined by qPCR and ELISA, respectively (Fig. 6H and Supplementary Fig. S7C). These data showed that the activation of the STING pathway by SR-717 administration can counteract the inhibitory effects on vascular endothelial inflammation caused by GSDMD blockade to some extent. Moreover, the phosphorylation levels of STING, TBK1, IRF-3, and p65 were substantially elevated with the introduction of SR-717 in the GSDMD-knockdown background. There were no differences in the protein levels of STING, TBK1, IRF-3, and p65 (Fig. 6I, J). These data indicate that the activation of the STING pathway by SR-717 can partially offset the inhibitory effects on the activation of the STING pathway. In summary, endothelial GSDMD plays a critical role in LPS-accelerated atherosclerosis via the positive regulation of the STING pathway in vivo.
We then treated GSDMD-knockdown HUVECs with SR-717 after treatment with ox-LDL plus LPS or the control. Our findings revealed that SR-717 administration can increase the protein expressions of ICAM1 and VCAM1 as determined by Western blot (Fig. 7A, B) and increase the mRNA expression levels of IL-1β, IL-6, IL-18, and MCP-1 mRNA levels as determined by qPCR (Fig. 7E). Moreover, the increase in IL-1β, IL-18, and LDH release levels in supernatants of the culture medium due to SR-717 was proved by ELISA and LDH assay (Supplementary Fig. S9B, D). The human THP-1 monocyte adhesion assay also revealed the increased adhesion abilities of endothelial cells (Fig. 7C, D). These results showed that the activation of the STING pathway by SR-717 could partially counteract the inhibitory effects on endothelial inflammation caused by GSDMD expression silencing. And evidently, elevated expression levels of p-STING, p-TBK1, p-IRF-3, and p-p65 were detected in GSDMD-knockdown HUVECs exposed to ox-LDL plus LPS or the control with SR-717, whereas the total protein expressions of STING, TBK1, IRF-3, and p65 remained unchanged (Fig. 7F, G). Our findings suggest that the activation of STING pathway by SR-717 can mitigate the inhibitory effects on the STING pathway activation in vivo.
To further investigate whether the activation of the cGAS-STING pathway is directly related to the release of mtDNA, we transfected HUVECs with the isolated mtDNA. Similar to the STING agonist SR-717, the transfection of mtDNA in HUVECs could have also resulted in the upregulation of phosphorylation levels of STING, TBK1, IRF-3, and p65, whereas the overall protein levels of these molecules remained unchanged (Supplementary Fig. S10A, B). We also discovered that the inhibitory effects on STING pathway activation resulting from silencing GSDMD expression can be partially reversed after mtDNA overexpression (Supplementary Fig. S10C, D).
Consistent with the findings of our animal experiments, we confirmed the importance of endothelial GSDMD in positively regulating the mtDNA-STING pathway during endothelial injury caused by long-term low-grade ox-LDL and LPS stimulation in vitro.
The transcription factor STAT3 promotes the transcription of the GSDMD gene
We performed bioinformatics analysis to predict the potential transcription factors of GSDMD on the basis of the GeneCards and JASPAR databases. Considering the intersection of the two databases, 15 genes were considered candidate transcription factors, including MYC, HEY1, ATF2, MAZ, ZNF384, CTCF, ELF3, ELF1, ZEB1, NR3C1, STAT3, RELA, NR2F2, TCF7L2, and EGR1. Multiple studies have revealed that STAT3 is involved in the development of atherosclerosis and sepsis (Wei et al., 2023; Zhao et al., 2016). We found that compared with no LPS stimulation, LPS-accelerated atherosclerosis led to greater STAT3 phosphorylation in the aortas of the 18-week-old ApoEKO mice (Fig. 8A, B). Compared with that in the control group, the phosphorylation of STAT3 increased in HUVECs exposed to each of the three intervention conditions, namely, ox-LDL, LPS, and ox-LDL plus LPS. The expression of p-STAT3 was greatest when HUVECs were exposed to the combination of ox-LDL and LPS (Fig. 8C, D). The trend of the variations in p-STAT3 was consistent with the expression of GSDMD. In this study, we hypothesized that STAT3 may be a potent transcriptional regulator of GSDMD. As expected, the protein expressions of GSDMD substantially decreased in STAT3-knockdown HUVECs (Fig. 8E, F). Furthermore, we predicted the conserved binding motif of STAT3 and several potential binding sites between STAT3 and the GSDMD promoter with the help of the JASPAR database (Fig. 8G, H). Notably, the regions from 611 bp to 621 bp and 234 bp to 244 bp upstream of the transcription start site of the GSDMD gene on human chromosome 8 appeared higher in the ranking order, and we performed mutagenesis of the two positions simultaneously for subsequent experiments. The results of dual-luciferase reporter assays showed that in human embryonic kidney-293 (HEK-293) cells transfected with pGL3-GSDMD-WT plasmids, STAT3 overexpression significantly increased the luciferase activity (Fig. 8I). Therefore, these data suggest that STAT3 is involved in the transcriptional regulation of GSDMD expression as a transcription factor.
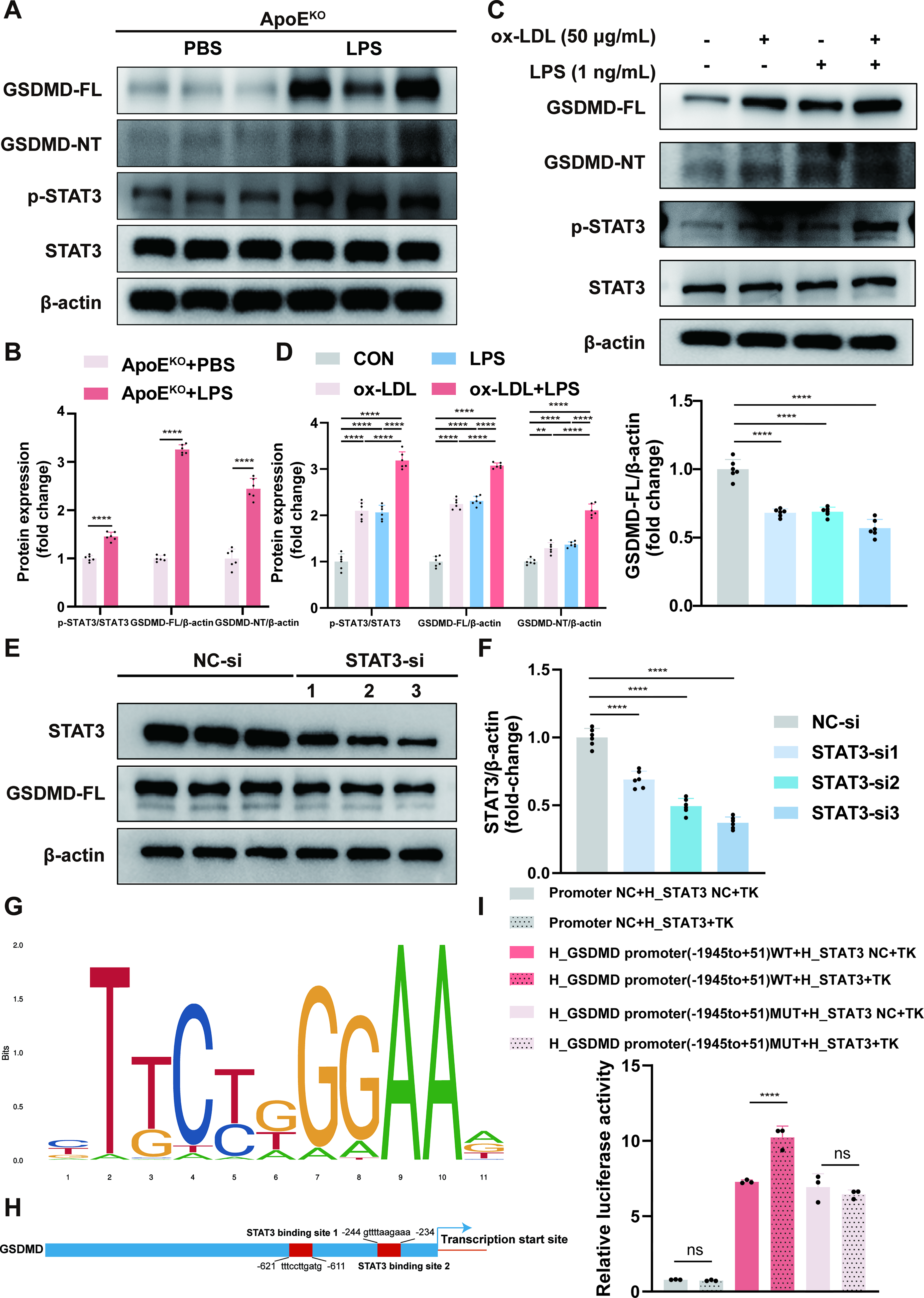
Taken together, these results indicate that endothelial GSDMD, which is regulated by expression and activation under exposure to chronic low-grade LPS, exerts its atherogenic effects by increasing mitochondrial damage and upregulating the activity of the STING pathway.
Discussion
Several studies have shown that chronic inflammation is an important risk factor for the onset and progression of cardiovascular and cerebrovascular diseases. Growing amounts of evidence support an important link between low-grade endotoxin-induced chronic inflammation and atherosclerosis (Suh et al., 2019; Wiedermann et al., 1999). Unlike acute inflammation, the sustained effects of systemic chronic inflammation result in significant physiological alterations in tissues and organs over time and increase the risk of noncommunicable diseases such as cardiovascular diseases (Furman et al., 2019). In this context, we focused on the mechanism of low-grade persistent LPS-accelerated atherosclerosis, and endothelial GSDMD may be a potential target for mediating the inflammatory response and mitochondrial damage during this process. These conclusions are supported by several in vivo and in vitro findings showing that: (1) Endothelial GSDMD expression in vivo and in vitro was upregulated in parallel with the exacerbation of atherosclerotic lesions and inflammatory responses. GSDMD deficiency attenuated the atherogenesis progression and vascular endothelial inflammation that was promoted by LPS in vivo and in vitro, and a STING agonist (SR-717) partially reversed the protective effects of GSDMD blockade. (2) By virtue of its pore-forming and mitochondrial localization properties, activated GSDMD induced the release of mtDNA into the cytoplasm to activate the STING signaling pathway, followed by the release of pro-inflammatory factors and the progression of atherosclerotic diseases. GSDMD ablation protected against the progression of mitochondrial damage, the release of mtDNA, and the activation of the STING pathway in vivo and in vitro. SR-717 administration partially reversed the protective effects of GSDMD blockade. (3) The expression of the transcription factor STAT3 was also upregulated in vivo and in vitro, and STAT3 expression silencing led to a reduction in GSDMD expression. STAT3 can directly bind to the GSDMD promoter to regulate its gene transcription.
To mimic the systemic chronic inflammation in vivo, we administered the weekly injections of LPS (0.6 mg/kg) based on the high-fat diet for 12 weeks by referring to the LPS dose (0.5 mg/kg) in a low-grade endotoxemia-related animal experiment (Carnevale et al., 2020). In a subclinical endotoxemia-complicated atherosclerosis cell model, the co-stimulation of ox-LDL (50 μg/mL) and LPS (1 ng/mL) induced macrophage inflammatory response (Wiesner et al., 2010). It has been proved that ox-LDL treatment (50 μg/mL) for 24 h can trigger endothelial inflammation (Liu et al., 2017). After evaluating the cooperative effects of LPS (0.1/1/10 ng/mL) plus ox-LDL (50 μg/mL) in the present study, we also found that the co-stimulation of ox-LDL (50 μg/mL) and LPS (1 ng/mL) significantly worsened the endothelial inflammation. The dose of LPS used corresponds with the findings from the Bruneck Study, in which low-grade endotoxemia subjects (with serum LPS levels beyond 50 pg/mL) faced a threefold risk of atherosclerotic events (Wiedermann et al., 1999).
GSDMD-mediated pyroptosis may have connections with atherosclerotic plaque burden and plaque stability (Puylaert et al., 2022; Shao et al., 2022). Mounting evidence suggests that pyroptosis during the progression of atherosclerosis can occur in a range of cells, including endothelial cells, smooth muscle cells, and macrophages (Qian et al., 2021). In our study, we discovered that administration of ox-LDL combined with low-dose LPS promoted the expression of ICAM1 and VCAM1 in endothelial cells and enhanced the capacity of endothelial cells to adhere to monocytes. These results indicated that the worsening of atherosclerosis by chronic exposure to low doses of LPS was related to endothelial injury. We observed elevated endothelial GSDMD expression and activation in LPS-accelerated atherosclerosis in the present study, and the colocalization of GSDMD with the endothelial marker CD31 was verified. Therefore, we aimed to provide insights into the role of endothelial GSDMD in LPS-accelerated atherosclerosis.
To investigate the function of GSDMD in vivo, we constructed GSDMD knockout ApoEKO mice and endothelial-specific GSDMD knockout ApoEKO mice. We found that GSDMD deletion alleviated atherogenesis progression, suggesting that endothelial GSDMD expression is involved in LPS-accelerated atherosclerosis. These results are in accordance with the results described in previous studies (Puylaert et al., 2022). Correspondingly, the knockdown of endothelial GSDMD in vitro significantly reduced endothelial cell inflammatory responses induced by chronic exposure to low doses of LPS. The abovementioned results indicated that endothelial GSDMD expression accelerates atherosclerosis development. Previous studies have shown that GSDMD expression is likely to exert adverse effects, such as worsening atherosclerosis by regulating the cross talk among apoptosis, autophagy, and pyroptosis, enhancing NETosis, inducing endoplasmic reticulum stress, and dampening reverse cholesterol transport (de Vasconcelos et al., 2020; Fidler et al., 2021; Huang et al., 2024; Opoku et al., 2021; Qu et al., 2022; Shi et al., 2021; Sollberger et al., 2018).
It has been confirmed that mitochondrial damage might be responsible for the progression of atherosclerosis (Ciccarelli et al., 2023). Researchers have reported that in macrophages isolated from human atherosclerotic plaques, loss of DNMT3A or TET2 function leads to decreased expression levels of mitochondrial transcription factor A, mtDNA release, and activation of the cGAS-STING pathway, thus providing more detailed data to support the close link between mtDNA and atherosclerosis (Cobo et al., 2022).
In addition, we found that the trends of the variations in Caspase-1/11 were consistent with the expression of GSDMD-full length and GSDMD-NT. Inflammatory Caspase-1/11 cleaves GSDMD to form GSDMD-NT, the pore-forming domain. GSDMD-NT binds most strongly to the cardiolipin and phosphatidylinositol phosphates included in mitochondrial and bacterial lipids and less strongly to the phosphatidic acids and phosphatidylserines on the mammalian cell membrane inner leaflets and does not bind to phosphatidylethanolamines or phosphatidylcholines on either plasma membrane leaflets (Liu et al., 2016). Consequently, GSDMD-NT binding not only is confined to the plasma membrane but also extends to the mitochondrial membrane, where it induces the excess mtROS production, MMP reduction, cytochrome C release, and mtDNA leakage (Platnich et al., 2018; Rogers et al., 2019; Zhao et al., 2023). The pore-forming activity of GSDMD-NT is known to be dependent on its oligomerization. Recently, mtROS and oxidized mtDNA were reported to play crucial roles in inducing GSDMD oligomerization (Evavold et al., 2021; Miao et al., 2023a). This may represent a potential amplifying loop to enhance the effects of GSDMD due to mitochondrial damage.
The pore formation and mitochondrial localization properties of GSDMD were thoroughly investigated in the present study, and our findings further corroborated the speculation that mitochondrial damage and mtDNA release are mediated by GSDMD expression. Specifically, we detected the colocalization of GSDMD and the mitochondrial marker TOMM20 by cellular immunofluorescence staining. After persistent exposure to low-grade ox-LDL plus LPS, mitochondrial damage was monitored by assessing the MMP, MPTP, and mtROS in vitro and the inhibition of GSDMD expression protected against mitochondrial damage. The increase in the amount of dsDNA released into the cytosol of endothelial cells from atherosclerotic arteries was confirmed by the immunofluorescence staining for dsDNA and CD31, and the inhibition of GSDMD expression suppressed the release of dsDNA. In addition, qPCR analysis suggested that cytoplasmic dsDNA was derived mostly from damaged mitochondria and that the inhibition of GSDMD expression reduced the amount of mtDNA released. Our findings provide more adequate data on LPS-related mitochondrial damage in endothelial cells (Huang et al., 2020). According to a recent study, cleaved GSDMD-NT translocates to the mitochondria and causes mitochondrial damage before cell membrane damage (Miao et al., 2023b).
Previous studies have shown that cytoplasmic dsDNA sensed by the DNA sensors cGAS and cGAS can catalyze the production of the novel second messenger cGAMP and subsequently activate the downstream STING pathway (Hopfner and Hornung, 2020). In concert with the results reported in the literature, our in vivo and in vitro studies revealed that the mtDNA release after mitochondrial damage increased the phosphorylation levels of STING, TBK1, IRF-3, and p65, leading to the production of pro-inflammatory cytokines and the progression of atherosclerosis. The ablation of GSDMD expression can attenuate the activation of the STING pathway and protect against the development of atherosclerosis, which is in agreement with the relationship between GSDMD and the STING pathway in cardiac hypertrophy (Han et al., 2022). Moreover, the activation of the STING pathway by SR-717 can partially counteract the protective effects on atherogenesis progression and vascular endothelial inflammation caused by GSDMD deletion. Similarly, in in vitro study, we revealed that the mtDNA overexpression by the mtDNA transfection can activate the STING pathway after GSDMD knockdown. These findings strongly suggest that the mtDNA-cGAS-STING pathway is positively regulated by GSDMD expression. Interestingly, the excess secretion of mtROS has also been reported to activate the cGAS-STING pathway (Zhang et al., 2023b). On the basis of the excess production of mtROS after persistent low-grade stimulation with ox-LDL plus LPS in our study, we postulated that endothelial GSDMD expression mediated the progression of LPS-accelerated atherosclerosis via the mtROS-mtDNA-cGAS-STING axis. In addition, an mtROS-dependent GSDMD-NT pore formation mechanism in mitochondria has been reported (Weindel et al., 2022). In this way, the excess secretion of mtROS leads to the formation of more pores in mitochondrial membranes, indicating the potential for positive feedback to aggravate mitochondrial damage, which warrants further study. Of note, the chronic low-dose LPS-accelerated atherosclerosis seen in our study is a type of systemic chronic inflammation characterized by the senescence-associated secretory phenotype (SASP), and the cGAS-STING pathway is considered as a major regulator of SASP (Furman et al., 2019; Victorelli et al., 2023). The abovementioned relationship between mitochondrial damage and the SASP provides theoretical support for our findings that chronic exposure to low doses of LPS upregulates the activity of the STING pathway by inducing endothelial mitochondrial damage and leading to the aggravation of atherosclerotic lesions.
Our results showed that the expression and activation of GSDMD played a vital role in mitochondrial damage in the process of chronic low-dose LPS-accelerated atherosclerosis. However, the precise regulatory mechanism underlying the upregulation of GSDMD expression is not completely understood. It has previously been reported that GSDMD expression is regulated by STAT1(Xiong et al., 2022). Notably, STAT3, from the same family, was confirmed to be related to endothelial dysfunction and inflammation in atherosclerosis (Chen et al., 2019). And ox-LDL can activate STAT1 and STAT3 and promote inflammatory and fibroproliferative effects in atherosclerotic plaques (Mazière et al., 1999). In addition, STAT3 is also well-known for its role in the control of responses to infectious diseases and inflammation (Xu et al., 2020; Zhao et al., 2016). Thus, STAT3 is a promising candidate transcription factor for GSDMD. Our data demonstrated that LPS-accelerated atherosclerosis activated STAT3 in vivo and in vitro in parallel with increased GSDMD expression, and that knockdown of STAT3 expression resulted in the downregulation of GSDMD expression. We engineered mutations in the two high-ranking predicted binding sites between STAT3 and the GSDMD promoter, and a dual-luciferase assay was subsequently performed. It was ultimately demonstrated that STAT3 could promote the expression of GSDMD, and the validity of the two predicted binding sites was indicated.
However, the present work has several limitations whereby the other mechanisms of mitochondrial damage caused by GSDMD activation and the specific molecular mechanism by which STAT3 promotes GSDMD expression have not yet been completely elucidated. The interactions among endothelial cells and different immune cells in the context of LPS-accelerated atherosclerosis await further investigation.
Materials and Methods
Animal experiments
GSDMDKO mice and GSDMD loxP (GSDMDfl/fl) mice were generated by the CRISPR-Cas9 system by GemPharmatech (Nanjing, China). ApoEKO mice and endothelial cell-specific Cre transgenic mice (Tie2cre/+) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). GSDMDKO/ApoEKO mice were generated by crossing ApoEKO mice with GSDMDKO mice. Endothelial GSDMD-deficient (GSDMDECKO) mice were obtained by mating Tie2cre/+ mice and GSDMDfl/fl mice. GSDMDECKO mice were then mated with ApoEKO mice to derive GSDMDECKO/ApoEKO mice. Mice were housed in standard specific-pathogen-free cages with a 12-h light/12-h dark cycle and fed on a high-fat diet (21% fat, 0.15% cholesterol, SLAC, Shanghai, China) from 6 to 18 weeks of age. All animal protocols were approved by the Shanghai Model Organisms Co., Ltd. Institutional Animal Care and Use Committee.
Male ApoEKO mice, GSDMDKO/ApoEKO mice, GSDMDWT/ApoEKO mice, and GSDMDECKO/ApoEKO mice were used as subjects for all the animal experiments. To mimic long-term low-grade inflammation, all the mice were challenged with intraperitoneal injections of low doses of LPS (0.6 mg/kg, L2880, Sigma-Aldrich, Missouri, USA) or PBS (G4202, Servicebio, Wuhan, China) once a week from 6 to 18 weeks of age. SR-717 (10 mg/kg, HY-131454, MedChem Express, Shanghai, China) or DMSO (HY-Y0320, MedChem Express, Shanghai, China) was administered daily to GSDMDWT/ApoEKO mice and GSDMDECKO/ApoEKO mice for the final 4 weeks of experiments.
Cell culture
HUVECs from the National Stem Cell Translational Resource Center (Shanghai, China) were cultured in endothelial cell medium (1001, ScienCell, California, USA) supplemented with 5% fetal bovine serum (FBS), 1% endothelial cell growth supplement, and 1% penicillin/streptomycin solution (P/S). The human monocytic THP-1 cell line and human embryonic kidney cell line (HEK-293) were from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). THP-1 cells were maintained in RPMI 1640 medium (KGM31800-500, Keygen, Nanjing, China) supplemented with 10% FBS (10270106, Thermo Fisher Scientific, Massachusetts, USA). HEK-293 cells were cultured in high-glucose DMEM (11995065, Thermo Fisher Scientific, Massachusetts, USA) supplemented with 10% FBS and 1% P/S (60162ES76, YEASEN, Shanghai, China). All the cells were grown in a humidified atmosphere at 37°C with 5% CO2.
To mimic long-term low-grade inflammatory effects in vitro, HUVECs were stimulated with 50 μg/mL ox-LDL (YB-002, Yiyuan, Guangzhou, China) plus 1 ng/mL LPS (tlrl-b5lps, InvivoGen, California, USA) for 24 h. SR-717 (3.6 μmol/L) was added to the medium 1 h before and during the costimulation of ox-LDL and LPS.
HE staining
The whole arteries from mice were fixed in 4% paraformaldehyde (G1101, Servicebio, Wuhan, China) for 4 h, dehydrated with 30% sucrose (A100335, Sangon, Shanghai, China) overnight, and embedded in optimal cutting temperature (OCT, 4583, Sakura, California, USA) compound at −40°C. The OCT compound-embedded frozen tissue was cryosectioned into 10-μm thick sections, and six sections with an interval of 80 μm were placed on a glass slide. The slides were stored at −80°C before use and recovered at room temperature (RT) for 1 h before staining. Then, the cryosections were stained with HE dye solutions, dehydrated in ethanol, cleared in dimethylbenzene, mounted with neutral balsam, and scanned with a Pannoramic scanner (3DHISTECH, Budapest, Hungary).
Immunofluorescence staining
For immunofluorescence staining of cryosections, the slides were kept at RT for 1 h and defatted in 95% ethanol and PBS. Then, the slides were permeabilized with 0.1% Triton X-100 (93443, Sigma-Aldrich, Missouri, USA) for 10 min and blocked in 0.1% Triton X-100, 5% bovine serum albumin (BSA, 36101ES60, YEASEN, Shanghai, China), 2% donkey serum (D9663, Sigma-Aldrich, Missouri, USA) for 30 min at RT. The slides were incubated with the following primary antibodies diluted in 0.1% Triton X-100–2.5% BSA-1% donkey serum overnight at 4°C: rabbit anti-GSDMD (NBP2-33422, Novus, Colorado, USA), rat anti-F4/80 (ab6640, Abcam, Cambridge, UK), goat anti-CD31 (AF3628, R&D Systems, Minneapolis, USA), and mouse anti-dsDNA (MAB1293, Millipore, Massachusetts, USA). The corresponding secondary antibodies were incubated for 2 h at RT: Alexa Fluor 594-conjugated donkey anti-rabbit (A-21207, Invitrogen, California, USA), Alexa Fluor 568-conjugated goat anti-rat (ab175476, Abcam, Cambridge, UK), Alexa Fluor 488-conjugated donkey anti-goat (A-11055, Invitrogen, California, USA), and Alexa Fluor 594-conjugated donkey anti-mouse (A-21203, Invitrogen, California, USA). Following washing with PBS, the slides were mounted with DAPI (P0131, Beyotime, Nanjing, China) and scanned with a Pannoramic scanner (3DHITECH, Budapest, Hungary).
For the immunofluorescence staining of cells, HUVECs were seeded into 6-well plates with 24-mm cell climbing slices during the intervention period. Cell fixation and permeabilization were performed before the blocking step. The cells were incubated with the following primary antibodies diluted in 0.1% Triton X-100–2.5% BSA-1% donkey serum overnight at 4°C: rabbit anti-TOMM20 (ab186735, Abcam, Cambridge, UK) and mouse anti-GSDMD (sc-81868, Santa Cruz, California, USA). After being washed with PBS, the cells were incubated with the following secondary antibodies for 1 h at RT: Alexa Fluor 488-conjugated goat anti-mouse (A-11001, Invitrogen, California, USA) and Alexa Fluor 555-conjugated goat anti-rabbit (A-21429, Invitrogen, California, USA). Following washing with PBS, the cells were counterstained with DAPI, and cell climbing slices were fixed on glass slides. Finally, representative images were acquired using a confocal laser scanning microscope (Leica, Heidelberg, Germany).
siRNA transfection
SiRNAs targeting GSDMD (GSDMD-si), STAT3 (STAT3-si), and scrambled siRNA were obtained from Genomeditech (Shanghai, China). HUVECs cultured in 6-well plates were transfected with these siRNAs (100 nmol/L) for 48 h using Lipofectamine 3000 (L3000015, Invitrogen, California, USA). After transfection with GSDMD-si, HUVECs were stimulated to establish cell injury models. Western blot analysis was performed to evaluate the transfection efficacy.
The sequences of the siRNAs for human GSDMD-si were as follows: sense 5′-GCAGGAGCUUCCACUUCUA-3′ and antisense 5′-UAGAAGUGGAAGCUCCUGC-3′. The sequences of the siRNAs for human STAT3-si were as follows: sense 5′-ACAUUCUUGGGAUUGUUGG-3′ and antisense 5′-CCAACAAUCCCAAGAAUGU-3′.
Western blot
Frozen tissues were homogenized and lysed in Radio Immunoprecipitation Assay lysis buffer (89900, Thermo Fisher Scientific, Massachusetts, USA) containing protease and phosphatase inhibitors (C600387 and C500017, Sangon, Shanghai, China) using a frozen grinding instrument (Servicebio, Wuhan, China). HUVECs were also immersed in the lysis buffer described above. The lysates were incubated on ice for 30 min and centrifuged at 12,000 g at 4°C for 30 min. Proteins in the supernatants were quantified using the Pierce bicinchoninic acid (BCA) Protein Assay Kit (23227, Thermo Fisher Scientific, Massachusetts, USA), separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride membranes, and blocked with 5% BSA before incubation with the following primary antibodies overnight at 4°C: mouse anti-GSDMD (sc-393656, Santa Cruz, California, USA), rabbit anti-GSDMD (NBP2-33422, Novus, Colorado, USA), rabbit anti-GSDMD (PU224937, Abmart, Shanghai, China), rabbit anti-pro Caspase-1+p10+p12 (ab179515, Abcam, Cambridge, UK), rat anti-Caspase-11 (NB120-10454, Novus, Colorado, USA), rabbit anti-p-STING (72971, Cell Signaling Technology, CST, Maryland, USA), rabbit anti-p-STING (AF7416, Affinity, Ohio, USA), rabbit anti-STING (13647, CST, Maryland, USA), rabbit anti-p-TBK1 (5483, CST, Maryland, USA), rabbit anti-TBK1 (3504, CST, Maryland, USA), rabbit anti-p-IRF-3 (29047, CST, Maryland, USA), rabbit anti-p-IRF-3 (29528–1-AP, Proteintech, Illinois, USA), mouse anti-IRF-3 (sc-33641, Santa Cruz, California, USA), rabbit anti-IRF-3 (DF6895, Affinity, Ohio, USA), rabbit anti-phospho (p)-NF-κB p65 (p-p65, 3033, CST, Maryland, USA), rabbit anti-p-p65 (TP56372, Abmart, Shanghai, China), rabbit anti-p65 (8242, CST, Maryland, USA), rabbit anti-p65 (F0006, Selleck, Shanghai, China), rabbit anti-p-STAT3 (9145, CST, Maryland, USA), rabbit anti-STAT3 (12640, CST, Maryland, USA), rabbit anti-ICAM1 (PB9018, and A00171, Boster, Wuhan, China), rabbit anti-VCAM1 (A01199 and BM4289, Boster, Wuhan, China), rabbit anti-MCP-1 (BA1843-2, Boster, Wuhan, China), rabbit anti-TOMM20 (ab186735, Abcam, Cambridge, UK), mouse anti-Histone H3 (68345–1-Ig, Proteintech, Illinois, USA), mouse anti-β-actin (BS6007MH, Bioworld, Nanjing, China), and mouse anti-GAPDH (MB001H, Bioworld, Nanjing, China). Following being washed with Tris-buffered solution with Tween-20, the membranes were incubated with HRP-labeled goat anti-rabbit/mouse IgG (Western blot [WB]0177 and WB0176, Weiaobio, Shanghai, China) for 2 h at RT. After washing, the bands were visualized using a ChemiDoc Touch Imager (Bio-Rad, California, USA), and the relative gray value of each band was quantified by ImageJ (NIH, Maryland, USA).
RNA extraction and real-time qPCR
Total RNA was extracted according to the instructions of the RNA Isolater Total RNA Extraction Reagent (R401-01, Vazyme, Nanjing, China). Reverse transcription was performed using Hifair III 1st Strand complementary DNA Synthesis SuperMix for qPCR (gDNA digester plus) (11141ES60, YEASEN, Shanghai, China). Real-time qPCR was conducted using ChamQ Universal SYBR qPCR Master Mix (Q711-02, Vazyme, Nanjing, China). The β-actin primers were purchased from Sangon (Shanghai, China), and the other primers used were from HuaGene (Shanghai, China). The qPCR primer sequences used are listed as follows: mouse IL-1β, forward primer 5′-TCGCAGCAGCACATCAACAAG-3′ and reverse primer 5′-TCCACGGGAAAGACACAGGTAG-3′; mouse IL-6, forward primer 5′-TTCTTGGGACTGATGCTGGTGAC-3′ and reverse primer 5′-CTGTTGGGAGTGGTATCCTCTGTG-3′; mouse IL-18, forward primer 5′-CTGGCTGTGACCCTCTCTGTG-3′ and reverse primer 5′-TGTCCTGGAACACGTTTCTGAAAG-3′; mouse MCP-1, forward primer 5′-ACTCACCTGCTGCTACTCATTCAC-3′ and reverse primer 5′-TCTTTGGGACACCTGCTGCT-3′; human IL-1β, forward primer 5′-CCGACCACCACTACAGCAAGG-3′ and reverse primer 5′-GGGCAGGGAACCAGCATCTTC-3′; human IL-6, forward primer 5′-TGGTGTTGCCTGCTGCCTTC-3′ and reverse primer 5′-GCTGAGATGCCGTCGAGGATG-3′; human IL-18, forward primer 5′-CTGGAATCAGATTACTTTGGCAAGC-3′ and reverse primer 5′-TCCTTGGTCAATGAAGAGAACTTG-3′; and human MCP-1, forward primer 5′-CGCTCAGCCAGATGCAATCAATG-3′ and reverse primer 5′-GATCACAGCTTCTTTGGGACACTTG-3′.
Mouse genotyping
Genotyping was carried out following the protocols of the One Step Mouse Genotyping Kit (PD101-01, Vazyme, Nanjing, China). Genomic DNA was extracted from the mouse toes, and the lysate was directly used as the template for PCR amplification. The products were fractionated by agarose gel electrophoresis, and the genotypes were identified. All the primers used were from HuaGene (Shanghai, China).
The primers used for genotyping the GSDMDKO mice were shown as follows: forward primer 5′-CGATGGAACGTAGTGCTGTG-3′ and reverse primer 5′-TCCTTCCCAACCTGCTGTTG-3′. For genotyping the GSDMDfl/fl mice, the primers used were shown as follows: forward primer 5′-TCTGTTCCCTCCAGCCCTACTTG-3′ and reverse primer 5′-CAGCAACCACAGCACTACGTTC-3′. For genotyping the Tie2-Cre mice, the primers used were shown as follows: forward primer 5′-CGCATAACCAGTGAAACAGCATTGC-3′ and reverse primer 5′-CCCTGTGCTCAGACAGAAATGAGA-3′. Three primers were used for genotyping the ApoEKO mice: forward primer 5′-GCCTAGCCGAGGGAGAGCCG-3′, wild-type primer 5′-TGTGACTTGGGAGCTCTGCAGC-3′, and reverse primer 5′-GCCGCCCCGACTGCATCT-3′.
Monocyte-endothelial cell adhesion assay
HUVECs were plated in 24-well plates and subjected to the abovementioned treatments. THP-1 cells (1 × 106 cells/mL) were resuspended in serum-free RPMI 1640 and labeled with 3 μmol/L Calcein AM (C2012, Beyotime, Nanjing, China) for 30 min at 37°C. Then, the THP-1 cells were washed with PBS and finally resuspended in a fresh medium. The labeled cells were added to the adherent HUVECs, incubated at 37°C for 1 h, and then rinsed with PBS to wash away the nonadherent cells. Finally, fluorescence images were captured with a fluorescence microscope (Leica, Heidelberg, Germany), and the number of adherent cells was counted in 10 random fields of each group using ImageJ (NIH, Maryland, USA).
Cell component fractionation and mtDNA quantification
Cell fractions were isolated using the Cell Fractionation Kit (ab109719, Abcam, Cambridge, UK). HUVECs were digested using 0.25% Trypsin-EDTA and collected by centrifugation. The cell pellets were resuspended in Buffer A and then mixed with Buffer B. After centrifuging the samples at 5000 g at 4°C for 1 min, we harvested several cell pellets and centrifuged the supernatant fractions at 10,000 g for 1 min. The supernatants obtained were the cytosolic fractions [C]. The two-step cell pellets were then resuspended in Buffer A and mixed with Buffer C. Following centrifugation of these samples at 5000 g at 4°C for 1 min, we harvested several cytoplasm-depleted cell pellets and centrifuged the resulting supernatant fractions at 10,000 g for 1 min. The supernatants obtained were the cytosolic fractions and mitochondrial fractions [M]. The two-step cytosol- and mitochondria-depleted cell pellets, which represented the nuclear fractions [N], were resuspended in Buffer A. Proteins in the supernatants were quantified using the Pierce BCA Protein Assay Kit (23227, Thermo Fisher Scientific, Massachusetts, USA). Then mix one volume of 5× SDS-PAGE Sample Buffer with four volumes of cytoplasmic, nuclear, and mitochondrial fractions, respectively. Total cytosolic DNA was extracted using a DNeasy Blood & Tissue Kit (69504, QIAGEN, Maryland, USA). For the quantification of cytosolic DNA, qPCR was conducted as described in our previous methods, and the ratio of mtDNA to nDNA was calculated to assess the cytoplasmic mtDNA release. All the primers used were from HuaGene (Shanghai, China). The qPCR primer sequences were shown as follows: human ND1, forward primer 5′-ATACCCATGGCCAACTCCT-3′ and reverse primer 5′-GGGCCTTTGCGTAGTTGTAT-3′; and human β-globin, forward primer 5′-GTGCACCTGACCTCTGAGGAGA-3′ and reverse primer 5′-CCTTGATACCAACCTGCCCAG-3′.
mtDNA isolation and transfection
Cell Mitochondrial DNA Isolation Kit (12021, Saint-Bio, Shanghai, China) was used to isolate mtDNA from stimulated HUVECs as the prescribed protocol. HUVECs cultured in 6-well plates were transfected with these mtDNA (1 μg/well) for 24 h using Lipofectamine 3000.
MitoSOX staining
HUVECs from different treatment groups were incubated with 5 μmol/L MitoSOX Red Mitochondrial Superoxide Indicator (40778ES50, YEASEN, Shanghai, China) for 10 min at 37°C. After being washed with warm HBSS buffer and counterstained with Hoechst 33342, the cells were observed under a fluorescence microscope (Leica, Heidelberg, Germany).
TMRE staining
HUVECs from different treatment groups were incubated with TMRE (C2001S, Beyotime, Nanjing, China) for 30 min at 37°C. After being washed with a warm medium and counterstained with Hoechst 33342, the cells were observed under a fluorescence microscope (Leica, Heidelberg, Germany).
JC-1 staining
HUVECs from different treatment groups were incubated with JC-1 (C2003S, Beyotime, Nanjing, China) for 20 min at 37°C. After being washed with staining buffer, the cells were observed under a fluorescence microscope (Leica, Heidelberg, Germany).
MPTP opening assay
According to the instructions of an MPTP Assay Kit (C2009S, Beyotime, Nanjing, China), HUVECs from different treatment groups were incubated with Calcein AM combined with CoCl2 for 30 min at 37°C and then immersed in fresh medium for an additional 30 min. After being washed with PBS, the cells were observed under a fluorescence microscope (Leica, Heidelberg, Germany).
ELISA assay
The levels of IL-1β and IL-18 in the serum of mice or the HUVECs supernatants were quantified using an ELISA assay kit (EK201B, EK218, EK101B, EK118, MultiSciences, Hangzhou, China). The ELISA assay was performed according to the manufacturer’s protocol.
LDH release assay
The LDH release assay was performed using an LDH Cytotoxicity Assay Kit (C0017, Beyotime, Nanjing, China) according to the manufacturer’s instructions. Briefly, the supernatants from untreated and treated HUVECs were collected and centrifuged at 400 g for 5 min. And a mixture composed of 120 μL of supernatant and 60 μL of LDH working solution was incubated in darkness at RT for 30 min, after which the optical density was measured at 490 nm. The formula involved is as follows: LDH release (%) = (Control or treated sample OD490−Background OD490)/(Maximum Enzyme Activity Control OD490−Background OD490) ×100%.
Dual-luciferase reporter assay
The pGL3-Basic vectors with wild-type GSDMD promoter sequences (−1945 to +51bp) or deletion mutant GSDMD promoter sequences (−1945 to +51 bp) luciferase reporter plasmids were co-transfected with STAT3 overexpression plasmids or control plasmids into HEK-293 cells at 70% confluence using HG transgene transfection reagent (TG-10012, Genomeditech, Shanghai, China) for 48 h. Luciferase activity was monitored by a dual-luciferase reporter assay system (GM-040502A, Genomeditech, Shanghai, China).
Statistical analysis
All the experimental data were analyzed and graphed using GraphPad Prism (version 8.0, GraphPad Software, California, USA). The data were shown as mean ± standard error of the mean. Statistical significance was analyzed with Student’s t-test for two-group comparisons and one-way ANOVA with Turkey post hoc test for multiple comparisons. Values of p < 0.05 were considered significant.
Authors’ Contributions
X.S.: Conceptualization, methodology, experimentation, writing—original draft, writing—review and editing, supervision. J.X.: Methodology, experimentation, writing—review and editing. E.S.: Methodology, experimentation, writing—review and editing. S.X.: Experimentation. X.C.: Experimentation. P.Y.: Writing—review and editing. L.W.: Writing—review and editing. M.L.: Conceptualization, supervision. H.J.: Conceptualization, supervision. All the authors read and approved the final article.
Footnotes
Author Disclosure Statement
We have no conflicts of interest to disclose.
Funding Information
This work was supported by grants from the National Natural Science Foundation of China (Grants Nos. 8197020946 and 82100392).
Ethics Approval
All the animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the Shanghai Research Center for Model Organisms. All experimental procedures involving animals were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Data Availability
Data will be made available on request.
Supplemental Material
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.