
Abstract
Randomized controlled trials (RCTs) are essential for the approval of new therapies; however, because of their design, they provide little insight concerning disease epidemiology/etiology and current clinical practice. Particularly, in lung disease, rigid inclusion/exclusion criteria can limit the generalizability of pivotal trial data. Noninterventional studies (NIS), conducted through the well-established mechanism of patient registries, are undervalued as a means to close data gaps left by RCTs by providing essential data that can guide patient care at different levels from clinical decision-making to health-care policy. While NIS contribute valuable data in all disease areas, their importance in rare diseases cannot be underestimated. In respiratory disease, registries have been essential in understanding the natural history and different phenotypes of rare conditions, such as alpha 1 antitrypsin deficiency, cystic fibrosis, and idiopathic pulmonary fibrosis. Importantly, additional therapeutic outcome data were generated. While measures for enhancing data quality in RCTs have evolved significantly, the approach and effectiveness of registries is variable. Within this article, we review the contribution of registries to pulmonary disease and make recommendations for their effective management. Additionally, we assess limitations of registry data as well as challenges to registry operation, including the impact of the European Union General Data Protection Regulation.
Introduction
Data from randomized controlled trials (RCTs) are considered the gold standard for evaluating the efficacy of an intervention and building evidence to support regulatory approval and drug licensing. Noninterventional studies (NIS), which comprise a group of studies with a variety of methodologies, can be used to collect real-word clinical data and to support findings of RCTs. In addition, NIS, often conducted through the well-established mechanism of patient registries, provide longitudinal outcomes, context in clinical practice, and help to shape future study initiatives. NIS are also frequently used to investigate the effects of drugs and other interventions but have been criticized for their potential to overestimate treatment effects, particularly if treatment is not concealed and a control group is lacking. 1 However, when analyzing the outcomes of single RCTs and observational studies or the conclusions of meta-analyses, the treatment effects reported in RCTs and observational studies were found to be very similar. 1,2 Therefore, NIS, particularly registry studies, provide an alternative source of data that can support both regulatory and routine clinical decision-making but are currently underutilized for this function. The purpose of this article is to assess the contribution of registry studies to the understanding of pulmonary diseases and outline requirements for the effective management and continuous improvement of registries in respiratory medicine.
Requirements for drug registration: The necessity of RCTs
Data from RCTs—particularly double-blind RCTs—are required to prove the efficacy of an intervention and are an integral part of the approval and licensing process for new therapies. RCTs are designed for success. This is achieved by reducing bias and confounding factors through precise study design and use of narrow inclusion and exclusion criteria, in addition to, the randomized allocation of study interventions. These measures increase internal validity 3 but come at a price: factors that create high internal validity in RCTs may reduce the generalizability of the data, with data on effects in broader populations often obtained postapproval. Low generalizability of RCT data has been particularly noted for common respiratory conditions. In a survey of 749 asthma patients, only 6% of those receiving asthma treatments were eligible to participate in RCTs of asthma medications, based on the Global Initiative for Asthma guidelines. 4 A further study found that by utilizing common inclusion criteria for clinical trials in chronic obstructive pulmonary disease (COPD), including nonatopic, forced expiratory volume in one second <70% predicted and smoking history >15 pack-years, fewer than 17% of the COPD population tested were eligible. 5
Pragmatic RCTs (pRCTs) utilize patient populations that closely mirror that of the general patient population and may increase the generalizability of clinical trials. 6 The differences in the approaches of RCTs and pRCTs, in addition to details of a recent high-profile example of a pRCT in respiratory medicine—The Salford Lung Study—are shown in Table 1. 7,8 Despite the advantages of pRCTs, they cannot be applied to all scenarios. A less homogenous study population and poor medication compliance contribute to increased variance in treatment response and a lower signal-to-noise ratio. 9,10 This can make it more difficult to determine significant differences between groups and may result in incorrect conclusions about the intervention. 9 Therefore, pRCTs do not fulfill regulatory guidelines and thus are not suitable for drug development.
Key differences between efficacy and effectiveness studies in comparison to the Salford Lung Study. 8

Source: Republished with permission of British Medical Journal, from Improving the reporting of pragmatic trials: an extension of the CONSORT statement, Zwarenstein M, et al. 337, 2008 7 ; permission conveyed through Copyright Clearance Center, Inc.
CAT: COPD assessment test; COPD: chronic obstructive pulmonary disease; EQ-5D: European Quality of Life–5 dimensions; LABA: long-acting beta agonist; LAMA: long-acting muscarinic agonists; pRCT: pragmatic randomized controlled trial; RCT: randomized controlled trial.
Challenges for RCTs in rare diseases
Regulatory agencies require data from RCTs before licensing a new compound in a rare disease. Although orphan drug legislation helps to facilitate drug development in rare diseases, conducting RCTs in this area poses additional challenges. 11,12 First, it is often difficult to recruit a large enough sample of comparable patients. 13 Eligible patients can be geographically dispersed worldwide. Second, trial recruitment can be hampered when a licensed drug is already on the market. Alpha 1 antitrypsin (AAT) therapy for the treatment of alpha 1 antitrypsin deficiency (AATD) has been on the market in some countries for a number of years, with US Food and Drug Administration (FDA) approval originally based on biochemical efficacy. 14,15 Consequently, a placebo-controlled trial to determine the clinical efficacy of AAT therapy in AATD (the RAPID clinical trial program) needed 4.5 years to recruit 180 patients worldwide. 16 In general, long recruitment and study periods provide further challenges. In the case of placebo-controlled studies, it may be ethically questionable to withhold treatment from patients for an extended period if there is already a good body of evidence for efficacy. In addition, long recruitment periods may jeopardize the study if advances in the quantification of disease severity have been made, for example, advancements in imaging technologies, or other drugs have become available. Patients may also choose to withdraw consent once a treatment becomes available in their country. Moreover, patient dispersal among treatment centers worldwide creates further issues. The inclusion of smaller centers with few patients and less experience of the disease within RCTs may add variability and bias, and RCTs could also be sensitive to patient variability because of environmental influence. Environmental factors may be particularly pertinent in respiratory disease, as different concentrations of ambient air pollutants could contribute to variations in baseline lung function and disease status/progression. 17 These factors may limit the utility of the outcome data recorded and can contribute to gaps within the final trial data.
A further complication for RCTs in rare disease arises when the natural history of a disease and the diversity of disease presentation are poorly understood. 18 This may mean that it is difficult to design the trial with regards to patient selection and duration of treatment/follow-up. In addition, where information concerning disease activity or disease progression is lacking, it can be difficult to select the most suitable clinical endpoint, potentially necessitating the use of a nonspecific outcome measure. 18,19 As a result, larger patient numbers and/or longer follow-up times may be required. This is particularly the case with patient-reported outcomes (PROs), for example, quality of life assessments. PROs are often less sensitive than other clinical outcomes and, due to their subjective nature, can be associated with higher interpatient variability. 20 For example, using St George’s Respiratory Questionnaire as an outcome parameter in AATD would require more than 8500 patients per study arm to be followed for more than 3 years to detect a 25% reduction in decline. 21 This is at a time when regulatory agencies are recommending that more pivotal studies provide data on PROs—the FDA recently set out to dramatically increase the level of patient input in clinical trials. 22
These factors mean that clinical trials in rare disease must be extremely well designed and highly controlled to maximize the potential to demonstrate significant differences between groups, and the difficulty in accurately assessing PROs in rare diseases represents a significant data gap. However, a recent article in the New England Journal of Medicine by Frieden et al. highlights that it may not always be the best course of action to wait for key data to be produced by an RCT when these data could be generated through well-designed and implemented NIS. 23 Therefore, in an ideal situation, results from both RCTs and NIS would be available for a disease, to provide complementary and unique insights into whom to treat, and with which treatment to obtain the best results.
Bridging the data gap with NIS
Clinical trials are characterized by an experimental design that randomly assigns patients to treatment arms or subgroups. In contrast, NIS observe and record the disease course and are designed to answer questions reflecting routine clinical practice and the impact of disease and treatments on patients’ everyday lives. Consequently, only treatments and diagnostic methods that are the normal standard-of-care can be used, and only marketed medical interventions can be utilized in accordance with their license restrictions. 24
NIS, including population, case-control, and cohort studies, each have their own unique features, aims, and study design (Figure 1). 25,26 Potentially, the NIS most frequently encountered in everyday clinical practice is the clinical audit, which is an essential tool for capturing a snapshot of health-care provision and highlighting areas for improvement. Although most frequently associated with local implementation, large-scale audits involving mass data collection that capture the status of service provision across countries and regions are becoming more frequent, the European COPD Audit being a key example. 27 However, for clinical research, and for investigating disease etiology and the effect of treatments in particular, longitudinal follow-up is required. An important type of NIS in this regard, and focus of the current review, is registry studies (Figure 2).


The utility of registry studies. AE: adverse event; PK: pharmacokinetics; PRO: patient-reported outcome; RCT: randomized clinical trial.
Bridging the gap: Focus on registry studies
Registry studies are inherently observational in nature, although some degree of control can be implemented in that any planned data analyses can drive the manner of the data collection. 28 The implementation of NIS provides an effective toolset for successful registries by facilitating longitudinal patient follow-up and uniform data collection. These data can be used to answer a broad range of questions that can contribute to strategic health-care planning, the development of disease prevention activities, improving health care quality, planning of clinical trials, regulatory policy, and clinical decision-making. 29 Although registries can contribute useful data in all disease areas, their use is essential in understanding the nature of rare diseases. Of particular importance are registries that include patients treated with Orphan drugs, as these databases can inform on the effectiveness and safety of therapies that received marketing authorization at a time when the clinical evidence was credible but not conclusive. 30 A summary of several important registries in respiratory medicine and their contributions to their respective fields are presented in Table 2. 31 –51
Examples of important registries in respiratory medicine and their contributions.

AAT: alpha 1 antitrypsin; AATD: alpha 1 antitrypsin deficiency; CF: cystic fibrosis; COPD: chronic obstructive pulmonary disease; GOLD: global initiative for chronic obstructive lung disease; IPF: idiopathic pulmonary fibrosis; PRO: patient reported outcome; QoL: quality-of-life; RCT: randomized controlled trial.
Considerations for the effective management of registries
Roadmap to the continuous improvement of registries
To maximize the usefulness of data produced by registries, high-quality data entry is essential. However, operating patient registries requires significant resources and organizational collaboration between many stakeholders; 13 yet independent rare disease registries, in particular, operate with few resources and often rely on the dedicated work of a few physicians.
Consequently, data quality in registries may be reduced due to inaccurate/incomplete data recording or incorrectly registered patients. 52 As a result, important disease outcomes can be inaccurately reported and the magnitude of any treatment effect can be under- or overestimated. To obtain good quality data and reduce the amount of bias introduced in the data collection, quality control should be similar to that employed in RCTs. Best practice for quality control in RCTs is based on the principles outlined in the International Council for Harmonization guidelines. 53 While there are guidelines on Good Epidemiological Practice, 54 a framework for data quality in registries, and to help ensure that data collection between centers is controlled and standardized, particularly when collecting data on PROs, is required. These principals are reflected in a recent discussion article by the European Medicines Agency’s Cross-Committee Task Force on Patient Registries, which has the objective of facilitating the use of patient registries in supporting regulatory decision-making. 55
When looking for guidance on the optimal operation of registries, it is also possible to look toward examples of effectively run registries. The US Cystic Fibrosis Foundation Patient Registry (CFFPR) has been invaluable in identifying the epidemiology of the genetic variants associated with the disease and monitoring the microbiology of chronic lung infections and the effectiveness of prophylactic antibiotics. The registry has been operational since 1986; in this time, it has accumulated data from over 48,463 CF patients. 56 Participation among patients assessed at CFFPR-accredited care centers is high, and few patients are lost to follow-up. A significant feature of the registry is the publication of annual summary reports that are freely accessible to the general public. These reports comprehensively review the status of CF diagnosis and management in the US, covering everything from patient demographics, diagnosis, and genetic testing to guidelines for screening and patient management, microbiology, current treatment modalities, and clinical outcome data. 47 The registry utilizes stringent quality assurance protocols to ensure that the data collected are comprehensive and highly accurate. A 2012 audit of the CFFPR demonstrated the rigorousness of data collection. Of patients included in the registry, data were recorded for 95% of clinic visits and 90% of hospitalizations. 56 In addition, all audited fields were found to be highly accurate, with inaccurate reporting ranging from 0.8% to 17.4%. The European equivalent to the CFFPR, European Cystic Fibrosis Society Patient Registry (ECFSPR), 49 has been similarly successful and is widely used for both academic and industry research purposes. Overall, the success of registries such as the CFFPR and ECFSPR is dependent on three criteria: quality of structure, quality of process, and quality of data (Table 3).
Prerequisites for a successful disease registry.
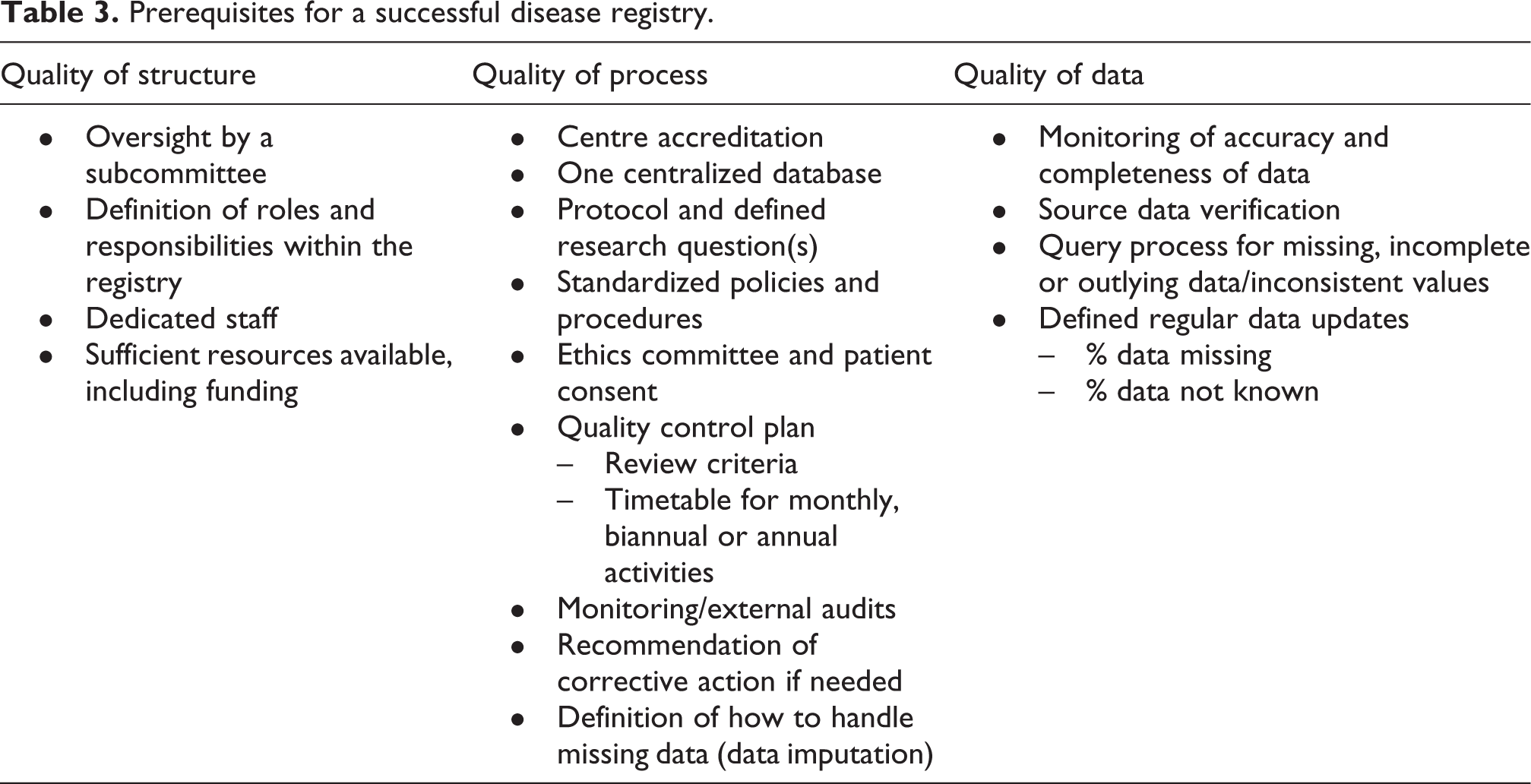
Quality of structure
A professional structure underpins the success of any registry. Many registries, particularly in rare diseases, are dependent upon the dedicated work of a small group of physicians. To generate the data output required to perform effective analyses in a timely and efficient manner, dedicated staff are required. In addition, oversight by a subcommittee and defined roles and responsibilities can ensure that funding structures are in place, research areas of interest are identified and focused on, and all regulations, such as data protection and good clinical practice guidelines, are adhered to. Quality of structure undoubtedly requires increased resources and funding; however, it can be argued that an efficiently run, well-resourced registry can contribute far more valuable data in the long term.
Quality of process
The processes of data collection and handling in registries are multifaceted. Standardization is an essential pillar that should be implemented within registries and agreed between international counterparts. This requires defined research protocols to ensure that methodologies do not deviate between centers contributing data. In addition, utilizing accredited centers (as in the CFFPR) can also aid the collection of a homogenous/comparable data set. A quality control plan and regular auditing can ensure that targets for data collection are met and that any deficiencies in standardization are addressed. Furthermore, effective design of the registry’s information technology infrastructure can contribute to quality assurance. The Alpha-1 Foundation research registry utilizes customized data archiving systems and portals for participating centers to achieve this. Another consideration is the ethical framework of the data collection, an extremely important aspect with potential legal ramifications if regulations are not adhered to. An ethics committee would usually be required to approve the conduct of any research studies and to ensure that all patient consent requirements are followed.
Quality of data
Inaccuracies in data quality, which can be due to a range of factors, including programing errors, transcription/typing errors, and deviations from data collection protocols, 57,58 can ultimately affect the outcome of any analyses performed. Procedures are therefore required for identifying outlying data and for recording and following-up on missing or incomplete data. To reduce imputation errors, databases can utilize automated logic check algorithms that prevent out-of-range values being entered and reject patient cases with missing/incomplete data—a system utilized by the European Multicentre Bronchiectasis Audit and Research Collaboration. 59 Overall, ensuring high-quality audited data is essential to secure stakeholder funding, particularly from industry.
Recognizing the limitations of registries and current challenges
Despite the advantages of registry studies described above, a number of limitations should be noted. A key strength of registry data is that it is more generalizable, as data input into registries is often not strictly monitored; however, the inclusion of ineligible patients, for example, those with less severe disease, can occur. 60 Furthermore, bias can be included at several stages, in part due to a lack of randomization in observational research (Figure 3). 61 Researchers should seek to limit the influence of biases wherever possible, for example, by ensuring accurate data collection and limiting selection bias. However, bias through confounding (both measured and unmeasured), which is largely mitigated by randomization in RCTs, is a particular issue for NIS. 62 It is therefore important for researchers to be aware of the statistical analyses that can be used to help adjust for confounders. The influence of measured confounders can be assessed through stratification and multivariate analyses (including logistic regression, linear regression, and analysis of covariance). 62 Unmeasured confounders, as would be expected, are more difficult to adjust for; however, several methods, such as the confounding function approach, have been developed in this regard. 63 The potential influence of unmeasured confounders in NIS highlights the importance of compressive data collection in the design of registries, and the level of evidence provided by registry studies is influenced to a large extent by the ability of analyses to adjust for confounders.

Types of bias in observational studies. 61
In addition to known limitations, open questions remain on how registry data can be utilized. The introduction of the new European Union General Data Protection Regulation (GDPR) may create significant issues for EU patient registries, particularly in rare diseases. In addition, although these directions only apply to EU citizens, any collaboration of US registries with EU counterparts would necessitate alignment with GDPR. GDPR lays down strict regulations that go beyond the current US confidentiality laws, that is, Health Insurance Portability and Accountability Act of 1996. 64 First, institutions involved in systematic data collection on a large scale that requires regular monitoring, such as patient registries, will be required to appoint a Data Protection Officer. 65 Second, there are further obligations to maintain documentation, implement data protection protocols, and conduct data protection impact assessments. 65 Third, the GDPR requires explicit informed consent separate from any other agreements; although existing consent agreements may still apply, it is likely that many will not. This has been a source of much controversy in the medical community, 66 particularly with respect to rare diseases. EPIRARE, the foremost European rare disease society, previously set up a petition to protest the requirements for new levels of consent that may have an adverse effect on the ability of registries to collect essential data. 67 The workability of these regulations within existing medical research apparatuses has yet to be determined; however, concessions for medical research may be required down the line to avoid these regulations having an adverse impact on essential data collection efforts.
In addition to data protection, data ownership is a major issue, which may be particularly pertinent within consortia-led registries. Public funding for patient registries is fiercely competed, and therefore, many rely upon fundraising activities or support from industry to fill this gap. Consequently, the funding of registries varies widely and there are often several stakeholders involved, including patients, patient advocacy groups, clinicians, academics, pharmaceutical companies, and government regulatory agencies and payers. 13 Some registries are fully maintained by government agencies—mandatory registries exist for significant threats to public health (e.g. tuberculosis), 68 and others are set up to encourage research, collaboration and increase knowledge and awareness on less-understood topics, the US Rare Disease Network being an example of this. 69 Alternatively, some groups receive funding from both industry and public bodies. The International Rare Disease Research Consortium—an extremely large worldwide network—is an example of this approach. 70 However, in scenarios where funding sources are mixed, it can be difficult to manage the expectations of all parties involved with regard to data collected and/or reported. To avoid controversy, detailed consortium agreements should be developed at the outset to ensure that the interests of each collaborating party are met. In addition, to maintain transparency, concerning any potential or perceived financial conflicts of interest, there is a need for protocols that define the scope of industry input and funding when setting up registries.
Conclusion
RCTs are essential for the approval of drugs; however, due to stringent control on trial recruitment and limited follow-up periods, the data often provide limited epidemiological/etiological insight, can lack generalizability, and can provide limited information on PROs. NIS, all of which can be conducted through patient registries, can be utilized to fill these data gaps. However, there are also limitations to the data generated by NIS, principally due to the lack of control for confounding factors. Therefore, RCTs and registry studies should be viewed in parallel, with both providing distinct but complementary data that helps our understanding of disease areas and treatment modalities. To obtain quality data from registries, there should be the same rigor in data collection and validation as applied to clinical trials. A well-designed registry should ensure the quality of structure, process, and data; this will undoubtedly require additional resources and dedicated staff. In addition, procedural requirements, including the new EU data protection regulations, should be carefully considered when establishing new patient registries. Overall, registry studies contribute to the understanding of disease epidemiology, clinical utility of new drugs, and policy making.
Footnotes
Authors’ note
JC-W reports grants, personal fees, and nonfinancial support from Grifols; personal fees from Kamada; grants, personal fees, and nonfinancial support from AstraZeneca; personal fees and nonfinancial support from Pfizer, MSD, and BMS; personal fees from GSK, Novartis, Takeda, and Chiesi; personal fees and nonfinancial support from Roche; grants and personal fees from Boehringer-Ingelheim; personal fees and nonfinancial support from AbbVe; grants, personal fees, and nonfinancial support from Celon Pharma; grants, personal fees, and nonfinancial from CSL Behring. MW reports personal fees from CSL Behring, GSK, and Sterna Biologicals. IH reports personal fees from AstraZeneca; personal fees and nonfinancial support from Boehringer-Ingelheim and Berlin-Chemie; personal fees from GSK; personal fees and nonfinancial support from Novartis, CSL Behring, and Roche; personal fees from Sandoz, Sager Pharma, Orion, and Affidea; personal fees and nonfinancial support from Teva; and nonfinancial support from MSD.
Acknowledgment
Medical writing assistance was provided by Steven Foster of Meridian HealthComms Ltd, Plumley, UK, in accordance with good publication practice (GPP3).
Author contributions
All authors contributed to the writing of the manuscript, reviewed the manuscript, and approved the manuscript for submission.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Medical writing assistance was funded by CSL Behring. CSL Behring are involved in the funding of registries in AATD, that is, the European Alpha-1 Research Collaboration (EARCO).