
Abstract
Sclerostin, a potent soluble inhibitor of the Wnt signalling pathway, is known to inhibit bone formation by suppressing osteocytes differentiation and function. Patients with chronic kidney disease have high levels of serum sclerostin. Sclerostin has been implicated in the pathogenesis of vascular calcification, which may promote the cardiovascular events of morbidity and mortality in chronic kidney disease patients. However, the role of sclerostin in vascular calcification and clinical prognosis in chronic kidney disease remains elusive. While some studies suggested a positive correlation between serum sclerostin and vascular calcification or clinical outcome, other studies showed no or even negative correlation between them. Small sample size, heterogeneity in enrolled patients, discrepancy in anatomical structure examined and differences in the applied assays may be responsible for the discrepant results. Nonetheless, anti-sclerostin antibodies may be a new therapeutic approach to increase bone mass and strength in chronic kidney disease. This review aims to have a better understanding of the relationship of serum sclerostin with vascular calcification and clinical outcome in chronic kidney disease patients, and propose the application of anti-sclerostin therapy in chronic kidney disease.
Introduction
Chronic kidney disease (CKD) has a high prevalence around the world, which contributes to huge medical costs and poses an enormous threat to public health. Cardiovascular problems are known to be the leading cause of mortality in CKD patients, 1 and the occurrence of cardiovascular events (CVEs) is remarkably higher in CKD patients than the non-CKD population. Abnormalities of mineral and bone metabolism and ectopic calcification, especially vascular calcification (VC), are common in CKD, resulting in a specific class of disease called chronic kidney disease-mineral and bone disorder (CKD-MBD). 2 CKD-MBD, which promotes the development of cardiovascular disease and gives rise to the increased risk of cardiovascular and all-cause mortality, is an extremely important complication of CKD, but it has often been neglected by clinicians. The pathogenic signalling pathway involved in this syndrome is still poorly understood.
Wnt/β-catenin signalling pathway participates in bone homeostasis and diseases.3,4 But beyond that, many lines of evidence derived from cell cultures and animal studies indicate that this signalling pathway plays a prominent role in the pathogenesis of atherosclerosis and VC.5–8 Sclerostin, a mainly osteocyte-derived soluble inhibitor of Wnt signalling pathway, is recognized as a potent inhibitor of bone formation by suppressing osteoblast differentiation, proliferation and promoting osteoblast apoptosis. 9 Cumulative evidence implies that serum or circulating sclerostin is higher in uraemic patients and serum sclerostin increases progressively across the CKD stages. However, the studies on the association of serum sclerostin with VC and mortality in renal disease patients have yielded conflicting results. Some investigations showed a positive correlation, whereas others suggested no or even negative correlation. Humanized monoclonal antibodies to neutralize sclerostin have been shown to increase bone mass and strength in postmenopausal osteoporosis women 10 and interestingly in animal models of progressive CKD.11,12
Sclerostin, an inhibitor of bone formation through Wnt/β-catenin signalling pathway
Sclerostin is a 22-kDa glycoprotein encoded by the SOST gene. This glycoprotein is almost exclusively synthesized by osteocytes that are not present near the bone surface but lie in the mineralized cortical and cancellous bone. Sclerostin can exert its inhibitory effects on bone formation by not only inhibiting proliferation, differentiation and function of osteoblast cells but also facilitating their apoptosis. 9 Sclerostin binds to the low-density lipoprotein (LDL) receptor-related family member LRP5/6, an important component of the Wnt signalling pathway, to inhibit the Wnt pathway resulting in the inhibition of bone formation.
Wnt signalling pathway participates in a wide range of physiological processes of embryonic development, including cell proliferation, differentiation and migration. The canonical Wnt signalling pathway starts by the binding of Wnt ligands to cell surface receptor complexes that are formed by transmembrane receptor Frizzled (FZD) and LRP5/6, which leads to the accumulation of β-catenin in the cytoplasm by a series of relay signals. Then, β-catenin can translocate into the nucleus where it interacts with and activates the transcription factor T-cell factor/lymphoid enhancer factor (TCF/LEF) to initiate the expression of target genes. Wnt/β-catenin pathway promotes bone formation by increasing the differentiation and function of osteoblast/osteocyte cells and by inhibiting osteoclast differentiation indirectly. 13 Sclerostin can directly bind to LRP5/6 to prevent binding of Wnt ligands to their receptors. As a result, sclerostin has notable inhibitory effects on bone formation. It has been recently acknowledged that VC is a process of gradual osteogenesis initiated by inflammatory factors in vessels. Furthermore, it has been documented that the Wnt signalling pathway can play a role in regulating endothelial inflammation, mesenchymal stem cell differentiation, and promoting the migration of monocytes, smooth muscle cells and endothelial cells.14–16 Additionally, it has been reported that missense mutations in LRP5 are associated with premature coronary artery disease. 17 Given the tight relationship between sclerostin and Wnt pathway, we speculate that the Wnt pathway may be an important player not only in the skeletal system but also in the vascular system through sclerostin.
Serum sclerostin levels in CKD patients are remarkably higher than those in the normal population, with their values increasing across the CKD stages. The increased sclerostin may be caused by the reduced renal clearance in CKD patients due to declining kidney function; however, this possibility is not supported by the recent work of Cejka et al. 18 Other investigators proposed that sclerostin expressed in the vascular system may release into the circulation and constitute a major portion of serum sclerostin, a view supported by the discovery of enhanced sclerostin expression in calcified vascular smooth muscle cells (VSMCs) and aortic valves. 5 Alternatively, the upregulated sclerostin levels may come from the increased production and secretion from bone; however, this view is challenged by a recent biopsy study that found no increase in bone sclerostin expression in the elderly whose serum sclerostin is enhanced. 19 To make a long story short, it is currently unclear whether the increased serum sclerostin is attributable to decreased renal clearance, excess skeletal/extraosseous production or both. A large amount of evidence has shown that ageing, male gender, diabetes, mechanical unloading of the skeleton, high serum LDL, low bone turnover, low parathyroid hormone (PTH) and low alkaline phosphatase (ALP) levels are associated with high sclerostin levels.9,20–22 Interestingly, besides these factors, the way of dialysis may also influence serum sclerostin level. In this regard, Lotte et al. 23 showed that compared with haemodialysis (HD), treatment with haemodiafiltration (HDF) can lead serum sclerostin levels to fall and the convection volume can alter the declining extent of serum sclerostin levels. Of note, there was no difference in serum sclerostin level between HD and peritoneal dialysis (PD) patients in this study.
Relationship between sclerostin and VC in CKD patients
VC occurs frequently in CKD patients and its incidence increases across the CKD stages. 24 Notably, VC is associated with CVEs and poor prognosis in CKD. It is well recognized that VC occurs in the early years of kidney disease patients recently, even prior to the occurrence of disordered phosphate homeostasis. 25 VC is recognized as a pathological process of osteogenesis initiated by inflammatory factors in vessels. However, the specific pathogenic mechanisms of VC are still poorly understood in CKD patients. VSMC transition to osteoblastic-like cells in the arterial tree is a well-appreciated mechanism of CKD-stimulated VC.26,27 In addition, reduction of vascular Klotho protein and upregulation of ALP and pyrophosphate hydrolysis may contribute to the development of VC.28,29 Enhanced expression of sclerostin was detected in calcified smooth muscle cells as well as in a mouse model of medial calcification. 5 Evidence has suggested that increased sclerostin may have a positive association with aortic calcification (AoC), abnormal intima-media thickness and carotid plaques in type 2 diabetes. 6 Hampson et al. 7 also reported similar positive correlation between plasma sclerostin level and abnormal AoC in postmenopausal women. Meanwhile, a significant upregulation of sclerostin messenger RNA (mRNA) was documented in calcified aortic valves of HD patients and in skin biopsies taken from HD patients with calciphylaxis. 8
The upregulation of sclerostin in calcified tissues led researchers and clinicians to come up with a hypothesis that sclerostin may be related to the pathogenic mechanism of VC in renal disease patients, leading to a large number of studies to examine the association between sclerostin and VC in CKD patients. However, these studies reported inconsistent results, with some studies showing positive association between sclerostin and VC in CKD patients,30–33 whereas others showing negative association34,35 or no association at all. 36 Qureshi et al. biopsied the inferior epigastric arteries of 89 newly transplanted patients and found that sclerostin can be used to predict the presence of VC and, even after their adjustment for confounding factors, this positive association persists. This study also pointed out that vasculature is not a primary source of serum sclerostin because they found no difference in serum sclerostin levels between calcified and non-calcified vessels. 32 Another study examined 161 stages 3–5 CKD patients including patients with dialysis by the abdominal lateral X-ray examination to evaluate aortic artery calcification (AAC) and, similar to the previous study, it found a positive correlation between the sclerostin levels and the presence of AAC. 33 It is worth noting that the sclerostin level was, however, found to be negatively associated with the severity of AAC in this study. 33 A possible explanation is that apoptosis of VSMCs is enhanced in patients with more severe AAC, which results in the decrease in overall production of sclerostin. As for the association between sclerostin and coronary artery calcification (CAC), Morena et al. 30 conducted research with chest multidetector computed tomography to assess CAC, involving 241 non-dialyzed CKD patients, and showed that after adjustment for confounders, the presence of CAC was positively associated with the sclerostin levels. In sharp contrast to these studies, Yang et al. 34 and Jean et al. 35 showed that serum sclerostin was negatively associated with AoC. Interestingly, three other studies37–39 suggested that sclerostin levels were positively associated with VC in univariate analysis, which became inverse in multivariate analysis. The detailed information and the results of related studies are listed in Tables 1 and 2. Based on these studies, we speculate that upregulation of sclerostin in the vascular wall may be a local negative feedback mechanism to inhibit the progression of VC.
Studies showing positive association between sclerostin and VC in CKD.
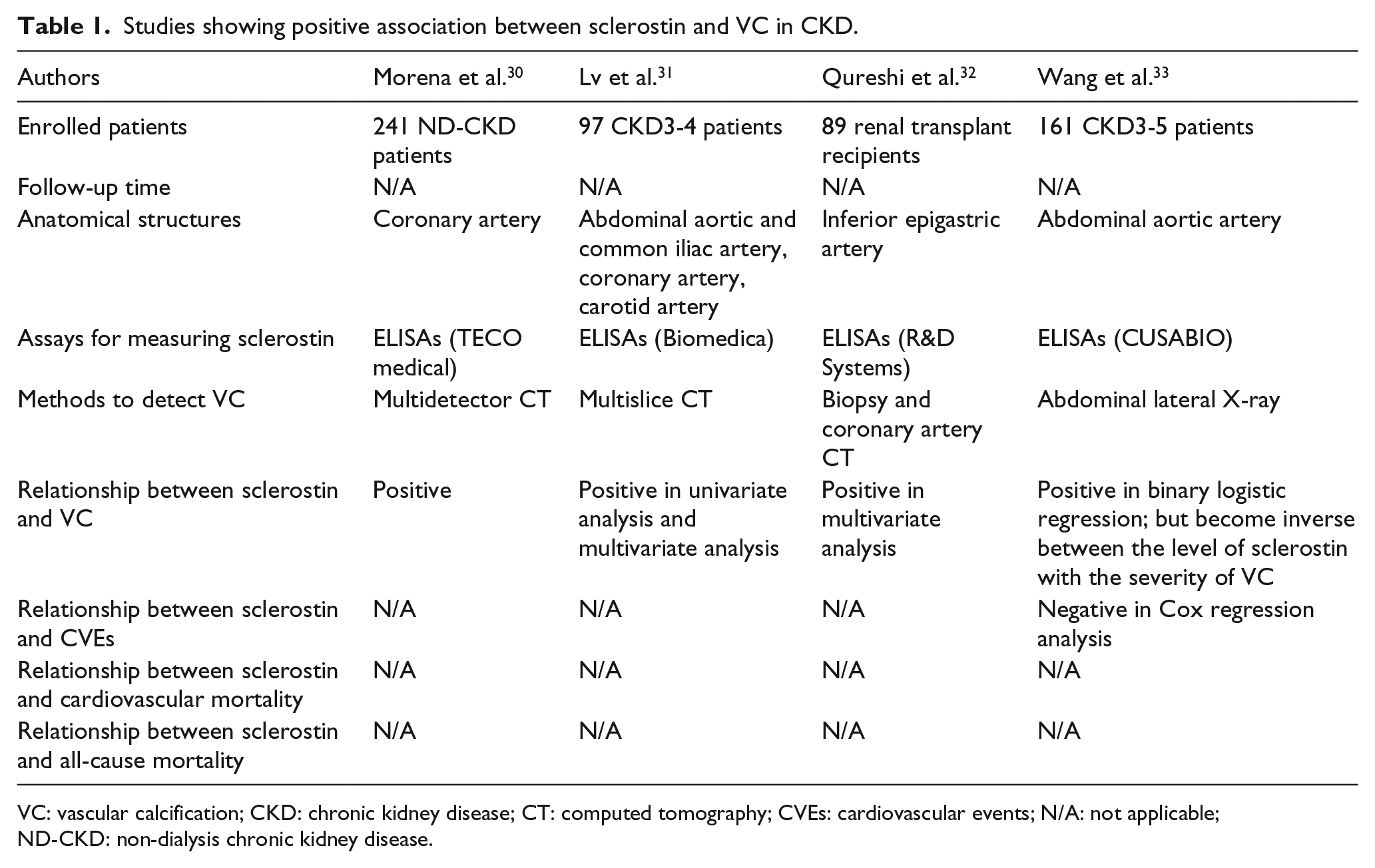
VC: vascular calcification; CKD: chronic kidney disease; CT: computed tomography; CVEs: cardiovascular events; N/A: not applicable; ND-CKD: non-dialysis chronic kidney disease.
Studies showing negative association between sclerostin and VC in CKD.
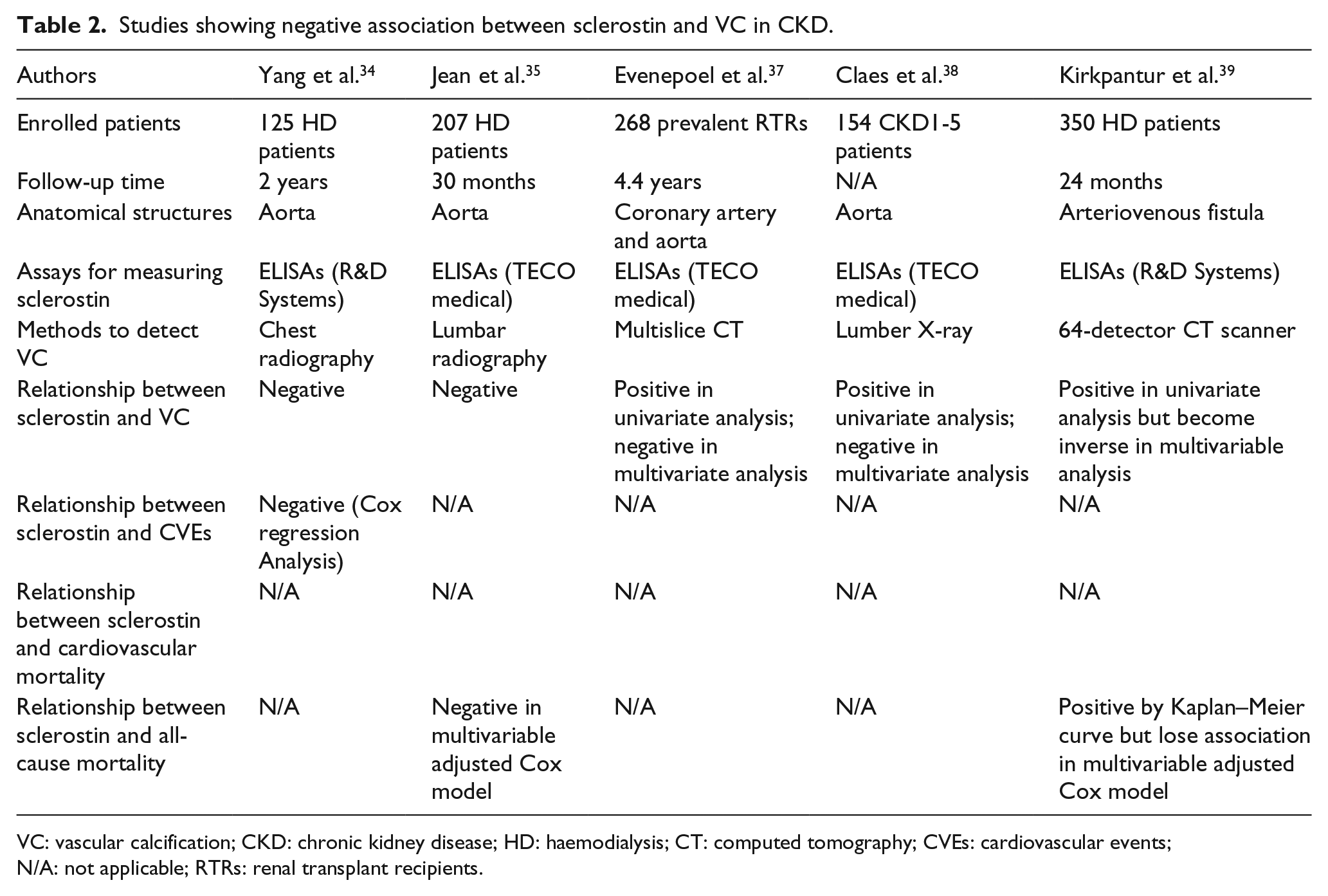
VC: vascular calcification; CKD: chronic kidney disease; HD: haemodialysis; CT: computed tomography; CVEs: cardiovascular events; N/A: not applicable; RTRs: renal transplant recipients.
Relationship between sclerostin and clinical outcome in CKD patients
The mortality in the end-stage renal disease patients remains unacceptably high, with a large portion of deaths ascribed to increased cardiovascular failure or dysfunction. Notably, this is associated with the high prevalence of traditional risk factors such as ageing, diabetes, hypertension and dyslipidaemia on a par with nontraditional risk factors including inflammation, oxidative stress and abnormal mineral metabolism.40–43 Concomitantly, fibroblast growth factor 23 (FGF-23), serum phosphorus and calcium, PTH levels and bone-specific ALP (bs-ALP, as biomarkers of mineral bone disorder) have been revealed to be associated with clinical outcome in renal disease patients.41–43 The syndrome of CKD-MBD includes the abnormalities of mineral bone metabolism and ectopic calcification, especially VC, which may lead to increased risk of cardiovascular and all-cause mortality. The various biomarkers mentioned above, which are related to the metabolism of mineral and bone, have been validated to be linked with the prognosis of kidney disease. Furthermore, sclerostin may participate in the pathogenesis of VC. Therefore, sclerostin, a known biomarker of bone formation, has drawn the attention of both researchers and clinicians for its potential association with cardiovascular and all-cause mortality in renal disease patients.
But, the results reported on the association between sclerostin and mortality are highly controversial. Five studies23,34,35,44,45 reported a decreased mortality and/or CVEs in the presence of higher serum sclerostin levels. Among them, the Netherlands Cooperative Study on the Adequacy of Dialysis (NECOSAD) study followed 673 dialysis patients (both HD and PD) for 18 months and showed that the risk of cardiovascular death and all-cause mortality were lower when sclerostin levels were higher after adjustment for a wide range of clinical and biochemical parameters including age, sex, estimated glomerular filtration rate (eGFR), blood pressure, levels of serum albumin, haemoglobin, intact PTH (iPTH) and ALP. Nevertheless, in this study, the correlation between sclerostin levels and cardiovascular mortality was less obvious for 4-year follow-up patients, and the association between sclerostin levels and non-cardiovascular mortality was absent. 45 Viaene et al. conducted a study of 100 dialysis patients, which detected the association of high sclerostin levels with improved survival after adjusting for age and gender. However, the association between sclerostin levels and clinical outcome lost statistical significance after full adjustment for bs-ALP; instead, higher bs-ALP was associated to worse survival. 44 Accordingly, Viaene et al. suggested that low bs-ALP activity may be the reason for patients with higher sclerostin having better survival. This may be explained by the ‘phosphate hypothesis’ where sclerostin may act as a suppressor of ALP to increase inorganic pyrophosphate (PPi), a potential local and circulating inhibitor of VC. Three studies38,46,47 showed that higher baseline levels of sclerostin were associated with worse survival. Kanbay et al. 47 examined 173 non-dialyzed CKD patients and 47 control patients to show that higher sclerostin levels, even after multiple statistical adjustments, were associated with fatal and non-fatal CVEs. However, it is worth noting that both Kanbay et al. 47 and Kirkpantur et al. 39 discovered that the relationship between serum sclerostin levels and all-cause mortality disappeared after multivariable Cox regression adjustment. In line with previous results, a meta-analysis of nine observational prospective studies demonstrated that circulating sclerostin levels are independent of all-cause and cardiovascular mortality. 36 The characteristics and the results of the studies mentioned above are listed in Tables 2 and 3.
Association between sclerostin and clinical outcome in CKD.
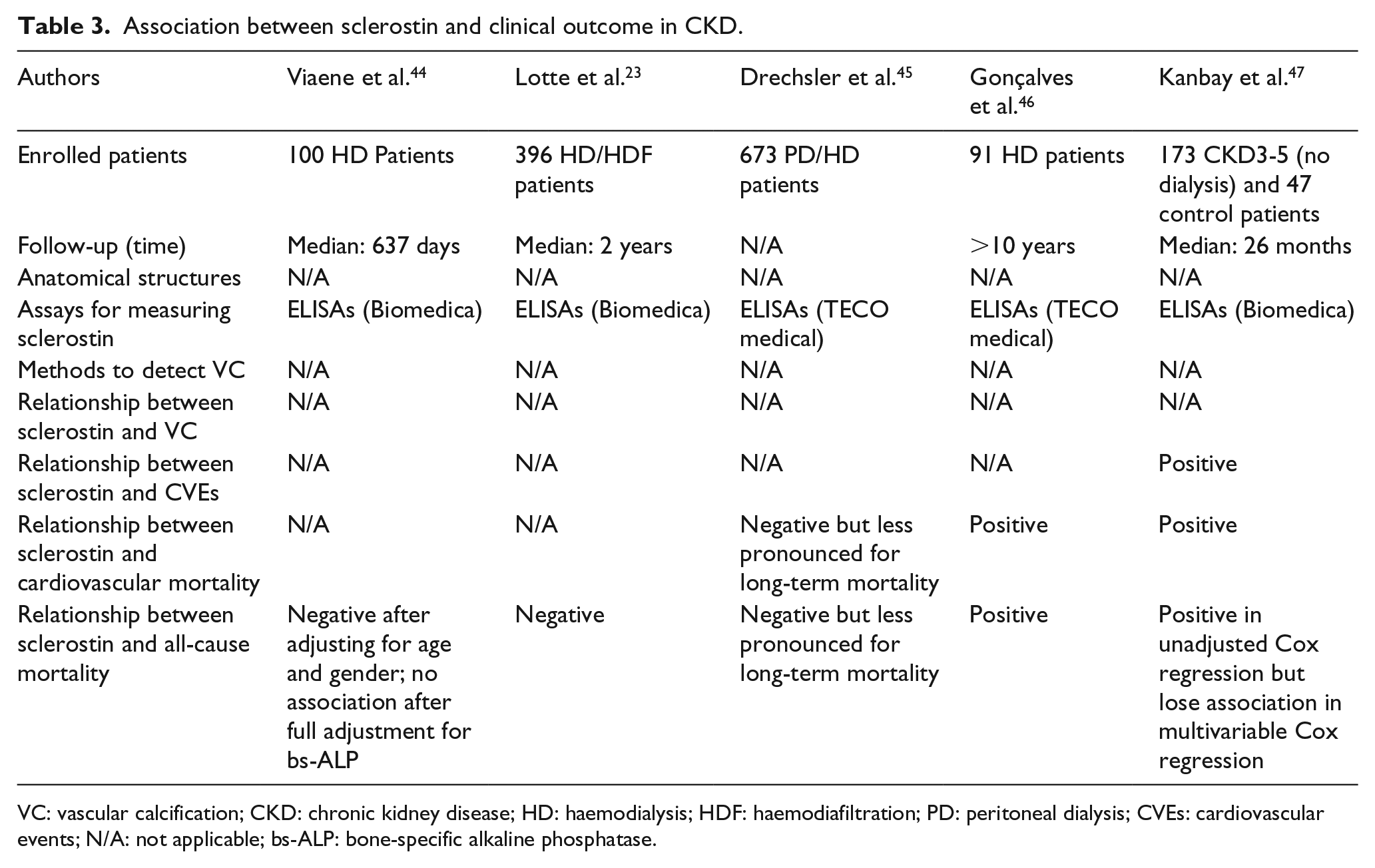
VC: vascular calcification; CKD: chronic kidney disease; HD: haemodialysis; HDF: haemodiafiltration; PD: peritoneal dialysis; CVEs: cardiovascular events; N/A: not applicable; bs-ALP: bone-specific alkaline phosphatase.
It is well established that sclerostin is emerging as a potent inhibitor of bone formation by reducing osteoblast differentiation and activity. We speculate that like in the bone, the upregulated sclerostin in the vascular wall may be part of the local defensive response to suppress further ossification. As a result, the attenuated progression of VC might be an explanation for the survival benefits in patients with higher sclerostin levels.
Therapeutic potential of anti-sclerostin antibodies in CKD
Sclerostin, a known inhibitor of Wnt signalling pathway, plays an indispensable role in the maintenance of normal bone quality. Two genetic diseases (sclerosteosis and Van Buchem disease), which are due to the absence of sclerostin expression, have significantly increased bone mass and decreased fracture rate. This observation led to the test of anti-sclerostin antibodies as new therapeutics to increase bone mass and strength. Three humanized monoclonal antibodies for neutralizing sclerostin (Scl-Ab), including romosozumab, blosozumab and BPS804, are in clinical investigation. The use of anti-sclerostin agents to neutralize sclerostin has been shown to be effective in increasing bone mineral density and bone strength in both animal models and clinical studies. The published clinical studies about the therapy of anti-sclerostin agents are mostly focused on treating osteoporosis. Renal osteodystrophy is an important complication in CKD-MBD and the patients with this complication suffer from increased incidence rates of fracture. But until now, there are no data on the application of anti-sclerostin agents in patients with CKD. Two preclinical studies that intend to assess the effectiveness of Scl-Ab in animal models of progressive CKD have been performed. Moe et al. 11 reported that compared to animals with high PTH, animals with low PTH can gain improved bone quality (e.g. through increasing the bone volume and trabecular mineralization surface) after the treatment of Scl-Ab. Newman et al. 12 also showed that anti-sclerostin agents can improve bone density and mechanical properties in another animal model of progressive CKD but only when used in combination with other therapy to normalize the PTH levels. A phase 1 clinical trial of romosozumab in stage 4 and stage 5 CKD patients has been completed (NCT01833754), but its results have not been published yet. Therefore, future studies are needed to clarify whether the use of Scl-Ab can improve bone quality in CKD patients with a sound safety profile and whether the employment of Scl-Ab can reduce cardiovascular morbidity and mortality in patients with CKD.
Conclusion
The relationship between sclerostin with VC and clinical outcome in CKD remains unclear. The published data in this area are highly inconsistent. The underlying causes of the inconsistence include small sample size, heterogeneity in enrolled patients cohorts (dialysis vs non-dialysis; HD vs PD), the discrepancy in anatomical structures examined (aortic valve, aortic artery and coronary artery), and the differences in the analytic methods for measuring serum sclerostin and calcification. Another possible explanation for the contradictory results is that sclerostin may have U-shaped effects on VC and mortality. In addition, the published studies are mainly associative and do not establish a causative role of sclerostin in VC and clinical outcome in CKD. Nonetheless, these studies have implicated sclerostin in the pathophysiological process of CKD-MBD. Numerous studies have confirmed that serum sclerostin is elevated in CKD although the source of the increase is still a subject of debate. As sclerostin antibodies are effective in increasing bone mass and strength in postmenopausal osteoporosis women and animal models of progressive CKD, it would be interesting to investigate the safety and the effectiveness of Scl-Ab in CKD-MBD patients.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
This study was supported in part by grants from the National Natural Science Foundation of China (81528004 and 81430017), the National Institutes of Health (DK058831, DK087843) and Department of Veterans Affairs of USA (BX000319).