
Abstract
Early studies showed nitric oxide as a pro-inflammatory-cytokine-induced toxin involved in pancreatic β-cell destruction during pathogenesis of type-1 diabetes. However, nitric oxide has both cytotoxic and cytoprotective effects on mammalian cells, depending on concentration and micro-environmental surroundings. Our studies have shown that low/physiological-level nitric oxide selectively activates protein kinase G type Iα isoform, promoting cytoprotective/pro-cell-survival effects in many cell types. In bone marrow–derived stromal/mesenchymal stem cells, protein kinase G type Iα mediates autocrine effects of nitric oxide and atrial natriuretic peptide, promoting DNA-synthesis/proliferation and cell survival. In this study, endothelial nitric oxide synthase/neuronal nitric oxide synthase inhibitor L-NIO (L-N(5)-(1-iminoethyl)ornithine), soluble guanylyl cyclase inhibitor ODQ (1H-[1,2,4]oxadiazolo[4,3,-a] quinoxalin-1-one), atrial natriuretic peptide–receptor inhibitor A71915 and protein kinase G type Iα kinase activity inhibitor DT-2 all increased apoptosis and decreased insulin secretion in RINm5F pancreatic β-cells, suggesting autocrine regulatory role for endogenous nitric oxide– and atrial natriuretic peptide–induced activation of protein kinase G type Iα. In four pancreatic β-cell lines, Beta-TC-6, RINm5F, INS-1 and 1.1B4, protein kinase G type Iα small-interfering RNA decreased phospho-serine-239-VASP (indicator of endogenous protein kinase G type Iα kinase activity), increased apoptosis and decreased proliferation. In protein kinase G type Iα–knockdown β-cell lines, expressions of phospho-protein kinase B (PKB/AKT) (AKT), phospho-Forkhead box protein O1 (FOXO1) (transcriptional repressor of pancreas duodenum homobox-1) and pancreas duodenum homobox-1 were decreased, suppressing proliferation and survival in pancreatic β-cells. The data suggest autocrine nitric oxide/atrial natriuretic peptide–induced activation of protein kinase G type Iα/p-AKT/p-FOXO1 promotes survival and proliferation in pancreatic β-cells, providing therapeutic implications for development of new therapeutic agents for diabetes.
Keywords
Introduction
Pro-inflammatory cytokines, produced in pancreatic islets during type-1 diabetes, stimulate high-level expression of inducible nitric oxide (NO) synthase (iNOS), generating NO locally in very high/toxic concentrations, and, when coupled with micro-environmental surroundings of elevated reactive oxygen species, contribute to β-cell death.1–6 High-level NO can cause β-cell death via its cytotoxic actions, altering protein functions via S-nitrosylation of cysteine residues and nitration of tyrosine residues,3,4,7–9 as well as by direct inhibition of cytoprotective enzymes, like catalase, as shown in RINm5F pancreatic β-cells. 10 Another proposed mechanism of high-level-NO-induced β-cell death is stimulation of soluble guanylyl cyclase (sGC)/cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG) signalling pathway. For example, in two β-cell lines, HIT-T15 cells 11 and BRIN-BD11 cells, 12 and isolated pancreatic islets from obese (ob/ob) mice, 13 high-level NO caused apoptosis via stimulation (likely pathological hyper-stimulation) of sGC/cGMP/PKG pathway. However, in other studies, NO at lower/more physiological levels did not cause apoptosis, but rather protected against apoptosis induced by serum starvation in RINm5F β-cells,14,15 and this cytoprotective effect appeared to be mediated by stimulation of an alternative/cytoprotective sGC/cGMP/PKG signalling pathway. Furthermore, exposure of INS-1 β-cells and rat pancreatic islets to atrial natriuretic peptide (ANP), a cardiac hormone that activates particulate guanylyl cyclase (pGC)/cGMP/PKG signalling pathway,16,17 stimulated DNA synthesis/cell proliferation via PKG’s ability to activate phosphatidylinositol 3′-kinase (PI3K)/p-AKT/p-FOXO1 pathway. 18
Notably, these earlier studies did not determine which isoform of PKG in pancreatic β-cells was involved in cytoprotective/proliferation responses to lower level NO and ANP. Our studies show that cytoprotective and DNA-synthesis/proliferation responses induced by physiological-level NO and ANP result from low-level stimulation of sGC/cGMP/PKG pathway involving an isoform of PKG (i.e. PKG-Iα (protein kinase G type Iα)) different from the one thought to mediate cytotoxicity. For example, physiological-level NO would selectively activate the PKG-Iα isoform, which is activated by relatively low concentrations of intracellular cGMP (half-maximal activation at 50–100 nM cGMP).7–9,19 In contrast, higher levels of NO, as would occur in inflammatory responses via pro-inflammatory-cytokine-induced iNOS expression or during hyperactivation of neuronal nitric oxide synthase (nNOS) or via high-level exposure to cGMP phosphodiesterase inhibitors (e.g. Exisulind), would additionally activate another isoforms of PKG-I, that is, PKG-Iβ.7–9,20,21 The PKG-Iβ isoform requires much higher concentrations of cGMP for activation (half-maximal activation at 1000 nM cGMP)9,19 and, when highly activated, is linked to cytotoxicity, resulting in inhibition of cell proliferation and induction of apoptosis.7–9,20
Supporting a cytoprotective role of lower level activation of sGC/cGMP/PKG pathway in pancreatic β-cells, exposure to cytoprotective-level carbon monoxide (a lower level activator of sGC/cGMP/PKG) protected βTC3 β-cells against tumour necrosis factor-α (TNF-α)-induced apoptosis and dramatically improved cell survival of transplanted pancreatic islet cells, significantly enhancing glucose-lowering effects of mouse pancreatic islets transplanted into diabetic mice. 22 Carbon monoxide, as a weaker sGC activator, would cause lower elevations of cGMP, thus selectively activating PKG-Iα isoform. Overall, low-to-moderate stimulation of sGC/cGMP/PKG pathway in pancreatic β-cells, likely involving selective activation of PKG-Iα, causes cytoprotection, reducing levels of apoptosis and improving cell survival and function.
Diabetes and obesity are known to damage physiological NO/cGMP/PKG pathway, including causing dramatic downregulation of PKG expression and/or kinase/catalytic activity in many types of mammalian cells. For example, cells exposed to elevated glucose levels or cells from diabetic animals and human patients with either type-1 or type-2 diabetes have much lower PKG kinase activity.23–25 Diabetes causes ‘NO resistance’, that is, inadequate biological responses to low/physiological-level NO, involving decrease in PKG kinase activity,24,25 contributing to insulin resistance and reduction in PI3K/AKT activation. 23 These studies show that diabetes-associated damage of PKG kinase activity contributes to cardiovascular complications and retinal damage. Also, erectile dysfunction (ED), often associated with diabetes, involves pathological decreases in PKG-I kinase activity, and, interestingly, ED in animal model is reversed by genetically induced replacement of PKG-Iα. 26 Although currently unknown, it appears likely that diabetes-induced suppression of PKG kinase activity also occurs in pancreatic β-cells and may contribute to damage of β-cell survival, proliferation and function.
Genetic ablation of PKG-I in mice, knocking out both PKG-I isoforms, induced a diabetes-like state, characterized by fasting hyperglycaemia and liver inflammation. 27 Thus, PKG-I expression is critical for keeping blood glucose levels under control and suppressing pro-inflammatory mediators that cause liver inflammation. This study also observed PKG-I tissue distribution and found high levels in liver stellate cells, but ‘undetectable levels’ of PKG-I in pancreatic β-cells, hepatocytes, fat cells and skeletal muscle cells (all potential targets of insulin action). A similar apparent lack of PKG-I in β-cells was observed in another study that found PKG-I immunoreactivity (by immunohistochemistry and conventional western blot analysis) only in α-cells and some δ-cells, but apparently not in β-cells, in pancreatic islets of mice. 28
Our studies have shown that PKG-I expression, at the protein level, is often difficult to detect in many cell types using conventional techniques. Unlike vascular smooth muscle cells (VSMCs) with high-level expression,19,29,30 other cells (e.g. endothelial cells and many cancer cells) have levels of PKG-I expression one-fifth to one-tenth of VSMCs, just at or below detection limits of conventional western blot analysis.7–9,30–34 Nevertheless, PKG-I expressed at these lower levels plays an important role in enhancing cell proliferation and survival of endothelial cells and cancer cells.8,9,33–35 In this study, we show that insulin-containing β-cells of mouse pancreatic islets and commercially available pancreatic β-cell lines, INS-1 (rat) and 1.1B4 (human), do indeed contain PKG-Iα/β protein, determined by immunofluorescent staining and imaging with confocal microscopy (see section ‘Results’).
The aims of this study are to (1) identify which PKG-I isoforms are expressed in pancreatic islets and pancreatic β-cell lines, using both conventional western blot analysis and ultrasensitive NanoPro 1000, a capillary isoelectric focusing (cIEF) technology with 1000-fold higher sensitivity and better protein-resolving capacity compared with conventional western blots; (2) investigate the role of endogenous NO/cGMP/PKG-Iα and ANP/cGMP/PKG-Iα signalling pathways in regulating cell survival/proliferation and insulin secretion in pancreatic β-cell lines, using both pharmacological inhibitors and small-interfering RNA (siRNA) gene knockdown targeting PKG-Iα expression; (3) determine whether PKG-Iα regulates other signalling pathways, such as AKT, FOXO1 and PDX-1 (pancreas duodenum homobox-1), major regulators of pancreatic β-cell phenotype, survival and proliferation. FOXO1 and downstream regulation of PDX-1 expression are recognized to play an important role in regulating pancreatic β-cell proliferation and survival 36 and were recently shown to involve in genetic alterations associated with the very high diabetes rate in Pima Indians of Arizona, USA. 37
Materials and methods
Cell cultures and reagents
OP9 mouse bone marrow–derived stromal/mesenchymal stem cells (BMSCs/BM-MSCs), RINm5F, PANC-1 and Beta-TC-6 cells were purchased from American Type Culture Collection (Manassas, VA, USA). The 1.1B4 cell line was purchased from Sigma–Aldrich (St. Louis, MO, USA). The INS-1 cell line was purchased from AddexBio (San Diego, CA, USA). BMSCs were cultured in α-Minimum Eagle’s Medium (α-MEM) and supplemented with 20% fetal bovine serum (FBS), streptomycin (50 µg/mL) and penicillin (50 units/mL). RINm5F, PANC-1 and Beta-TC-6 were maintained in RPMI-1640 medium with 10% FBS, streptomycin (50 µg/mL) and penicillin (50 units/mL) (Lonza, Walkersville, MD, USA). The 1.1B4 cells were maintained in RPMI-1640 medium (Gibco, Thermo Fisher Scientific, Carlsbad, CA, USA) containing 11.1 mM glucose and supplemented with 10% FBS, streptomycin (100 µg/mL) and penicillin (100 units/mL). The INS-1 cells were cultured in the same medium as the 1.1B4 cells except 0.05 mM 2-mercaptoethanol was added. Cells were cultured at 37°C in 5% CO2/95% air. L-NIO (L-N(5)-(1-iminoethyl)ornithine), ODQ (1H-[1,2,4]oxadiazolo[4,3,-a] quinoxalin-1-one) and A71915 were purchased from EMD Millipore (Billerica, MA, USA). DT-2 and DT-3 were purchased from Biolog (Hayward, CA, USA).
Primary pancreatic islet isolation
Adult male C57BL/6J mice (6–8 weeks old) from Jackson Laboratories (Bar Harbor, ME, USA) were used for isolation of primary pancreatic islets. Treatment of laboratory animals and experimental procedures of this study adhered to National Institutes of Health (NIH) Guidelines for Animal Care and were approved by IACUC at Nevada Cancer Institute, Summerlin/Las Vegas, NV, USA. Islets were isolated from pancreases. Mice were sacrificed by carbon dioxide, and pancreases were dissected and placed in cold Hank’s Balanced Salt Solution (HBSS, Lonza). Tissue was cut into small pieces and transferred to vials containing 8 mL digestion solution (3 mg/mL collagenase A, Roche Applied Science, Indianapolis, IN, USA). Vials were shaken vigorously at 37°C for 10 min. Digestion was terminated by addition of cold HBSS and washed three times. Islets were picked out using pipette and cultured in suspension for 24 h in non-adherent 35 mm culture dishes (Corning, NY, USA) in RPMI-1640 with 10% FBS (Gibco, Thermo Fisher Scientific, Carlsbad, CA, USA).
Immunofluorescent staining of dispersed islets
Approximately 100 mouse islets were dissociated into individual cells using Accutase, Cat# 1000449 (MP Biomedicals, Santa Ana, CA, USA) for 5 min at 37°C in water bath with gentle pipetting. Accutase activity was stopped by adding 5 mL of RPMI-1640 (10% FBS). Dissociated cell suspension was cytospined onto lysine-coated glass slides using cytocentrifuge (ELITech Biomedical Systems, Logan, UT, USA). Cells were fixed in 4% formaldehyde and permeabilized using 3% Tween-20, followed by incubation in primary antibodies for PKG-Iα/β (monoclonal Rabbit, Cat# 3248S, at 1:100; Cell Signaling Technology, Danvers, MA, USA) and insulin (Polyclonal Chicken, ab14042, at 1:500) (Abcam, Cambridge, MA, USA). Secondary antibodies were Anti-Rabbit Cy5 (Green in images) and Anti-chicken Alexa 546 (Red in images; Abcam). After final washing, slides were mounted using Citifluor, anti-fadent mounting medium (Cat# 17970-100) (VWR, Radnor, PA, USA). Imaging was done using Nikon’s A1R Confocal Laser Microscope (Nikon Instruments Inc., Melville, NY, USA).
Immunofluorescent staining of cell lines
Cells were fixed in 4% formaldehyde for 15 min at room temperature and permeabilized with 0.3% Tween-20 for 20 min at room temperature. The cells were then incubated in primary antibodies for PKG-Iα/β (rabbit monoclonal, Cat# 3248S, at 1:50; Cell Signaling Technology) and insulin (mouse monoclonal, Cat# sc-8033, at 1:50; Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C. After washing, the cells were incubated with donkey anti-rabbit TRITC (Cat# 711-025-152, at 1:50, green in images) and donkey anti-mouse Cy5 (Cat# 715-175-150, at 1:50, red in images) secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) for 30 min at room temperature. Imaging was done using Nikon’s A1R Confocal Laser Microscope (Nikon Instruments Inc.).
siRNA-mediated PKG-Iα knockdown
For siRNA-mediated silencing of gene expression, cells were transfected with 100 nmol/L siRNA Stealth RNAiTM (PKG-Iα siRNA 1, 5′-GAGGAAGACUUUGCCAAGAUUCUCA-3′ and PKG-Iα siRNA 2, 5′-CGACCUCCGACAGGCAUUCCGGAAG-3′) (Invitrogen, Carlsbad, CA, USA) for specifically targeting the expression of PKG-Iα, as described previously. 38 A second set of siRNAs was developed more recently for PKG-Iα knockdown and was performed by transfecting cells with 100 nmol/L or 200 nmol/L of siPKG-Iα S1 (5′-CAGGCAUUCCGGAAGUUCAdTdT-3′) or siPKG-Iα S2 (5′-GCCAGUCGGUGCUCCCAGUdTdT-3′) (Sigma–Aldrich) (St. Louis, MO, USA).
Assessment of apoptosis by Cell Death Detection ELISAPLUS
Apoptosis was measured by Cell Death Detection ELISAPLUS (Roche Applied Science) as described previously. 38
Assessment of de novo DNA synthesis by BrdU ELISA
DNA synthesis was measured by 5′-bromo-2′-deoxyuridine (BrdU) ELISA (Roche Applied Science) as described previously. 32
Assessment of cell proliferation by MTT assay
Proliferation was measured by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (Roche) as described previously. 34
Caspase-3/7 activity by chemiluminescence assay
Caspase-3/7 activity was determined using Caspase-3/7 Glo kit (Promega, Madison, WI, USA) as described previously. 38
Protein extraction and western blotting
Protein extraction/western blotting was performed as described previously. 38 Blots were probed with primary antibodies: PKG-Iα/β (1:1000), p-(ser239)-VASP (1:500), total vasodilator-stimulated phosphoprotein (VASP) (1:2000) (or 1:1000 for total VASP from Santa Cruz Biotechnology), p-(ser1177)-eNOS (1:1000), total endothelial nitric oxide synthase (eNOS) (1:1000), PDX-1 (1:1000), p-(ser256)-Foxo1 (1:500), total Foxo1 (1:1000), p-(thr308)-Akt (1:1000), p-(ser473)-Akt (1:1000), total Akt (1:1000), p-(ser133)-CREB (1:500), total cAMP response element binding protein (CREB) (1:1000), all from Cell Signaling Technology, β-actin (1:2000) (Santa Cruz Biotechnology), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:2000) (EMD Millipore) and subsequently with secondary antibodies labelled with infrared fluorescent dyes (LI-COR Biosciences, Lincoln, NE, USA) (1:25,000 in blocking buffer at room temperature for 1 h). In some cases, two proteins were probed on the same blot using two secondary antibodies labelled with two different fluorescent dyes. Membranes were scanned on Odyssey infrared imaging system (LI-COR Biosciences).
Co-culture system of BMSCs and RINm5F β-cells
BMSC OP9 cells (BMSCs/BM-MSCs) were used as a cell type previously shown to be involved in promoting regeneration of pancreatic β-cells 39 and also capable of synthesizing and secreting the cardiac hormone ANP as an autocrine pro-survival/pro-growth factor. 32 We tested whether co-culturing BMSC/OP9 with RINm5F cells enhances proliferation rate of RINm5F β-cells via BMSC release of ANP and subsequent activation of natriuretic peptide receptor type-A (NPR-A)/pGC receptors on RINm5F cells. BMSC/OP9 cells were grown in α-MEM with 20% FBS on top of 6-well Nunc© Anopore Polycarbonate Membrane Inserts with 0.2 µm pore size (Nalge Nunc International, Rochester, NY, USA). RINm5F β-cells were grown at the bottom of 6-well plates in Roswell Park Memorial Institute (RPMI) media with 10% FBS. During co-culture experiments, inserts containing BMSCs were placed into 6-well plates with growing RINm5F β-cells. Cells were maintained in reduced-serum RPMI media (1% FBS), and BrdU was added to media at time 0 with or without ANP receptor (NPR-A) antagonist A71915. RINm5F cells were harvested for BrdU assay at 72 h.
NanoPro 1000, a cIEF system for ultrasensitive identification of PKG-I isoforms in mouse islets and pancreatic cell lines
Methodology for NanoPro 1000 (ProteinSimple, Santa Clara, CA, USA) was described in detail previously.7,34,40,41 Briefly, cells were lysed with M-Per lysis buffer in presence of protease/phosphatase inhibitor cocktail, following recommendations from manufacturer. Cell lysates (62.5 ng total protein/µL) were prepared in Premix G2 pH 5–8 (nested) separation gradient containing pI standards and protease/phosphatase inhibitors. Separation time was 50 min at 15 mW. Anti-PKG-Iα/β antibody (Cell Signaling Technology), which recognizes both PKG-Iα and PKG-Iβ isoforms, was used as primary antibody, and anti-rabbit HRP (horseradish peroxidase) was used as secondary antibody. Incubations were carried out for 110 and 55 min, respectively. Chemiluminescent reagents were automatically introduced into capillaries of NanoPro 1000 system for generation of chemiluminescent signals. Results are expressed as electropherograms with peaks representing targeted protein and its various isoforms and phosphoforms.
Measurement of insulin secretion from RINm5F β-cells
RINm5F cells were seeded in 96-well plates. After treatment with inhibitors for 24 h, media were replaced with phenol-red-free/serum-free RPMI for 6 h. Media were collected and 20 µL per sample was used for measurement by insulin ELISA (EZRMI-13K; EMD Millipore).
Statistical analysis
Statistical analysis was performed by one-way analysis of variance (ANOVA) or a two-way ANOVA using Prism 5 software (GraphPad, San Diego, CA, USA). Differences between experimental groups were determined by Dunnett’s post-hoc test (one-way ANOVA) or Bonferroni posttest (two-way ANOVA) for comparison of multiple treated groups to a common control. A value of p < 0.05 was considered to be significant.
Results
Both pharmacological inhibitors and gene knockdown methodology were used to define the role of NO/cGMP/PKG-Iα and ANP/cGMP/PKG-Iα signalling pathways in regulating apoptosis, cell proliferation rates and insulin secretion in pancreatic β-cells. A model showing cellular mechanism and sites of inhibition is illustrated in Figure 1(a). We used inhibitors that target each of the key steps in the signalling pathways, including the eNOS/nNOS inhibitor L-NIO, the sGC inhibitor ODQ, the ANP receptor (NPR-A/pGC) inhibitor A71915 and the PKG-Iα inhibitor DT-2 (also DT-3), 42 as well as PKG-Iα-specific siRNA to knockdown PKG-Iα gene expression. We also identified the isoforms of PKG-I expressed in pancreatic islets and various pancreatic cell lines and further determined whether PKG-I is located within insulin-containing β-cells of mouse pancreatic islets.
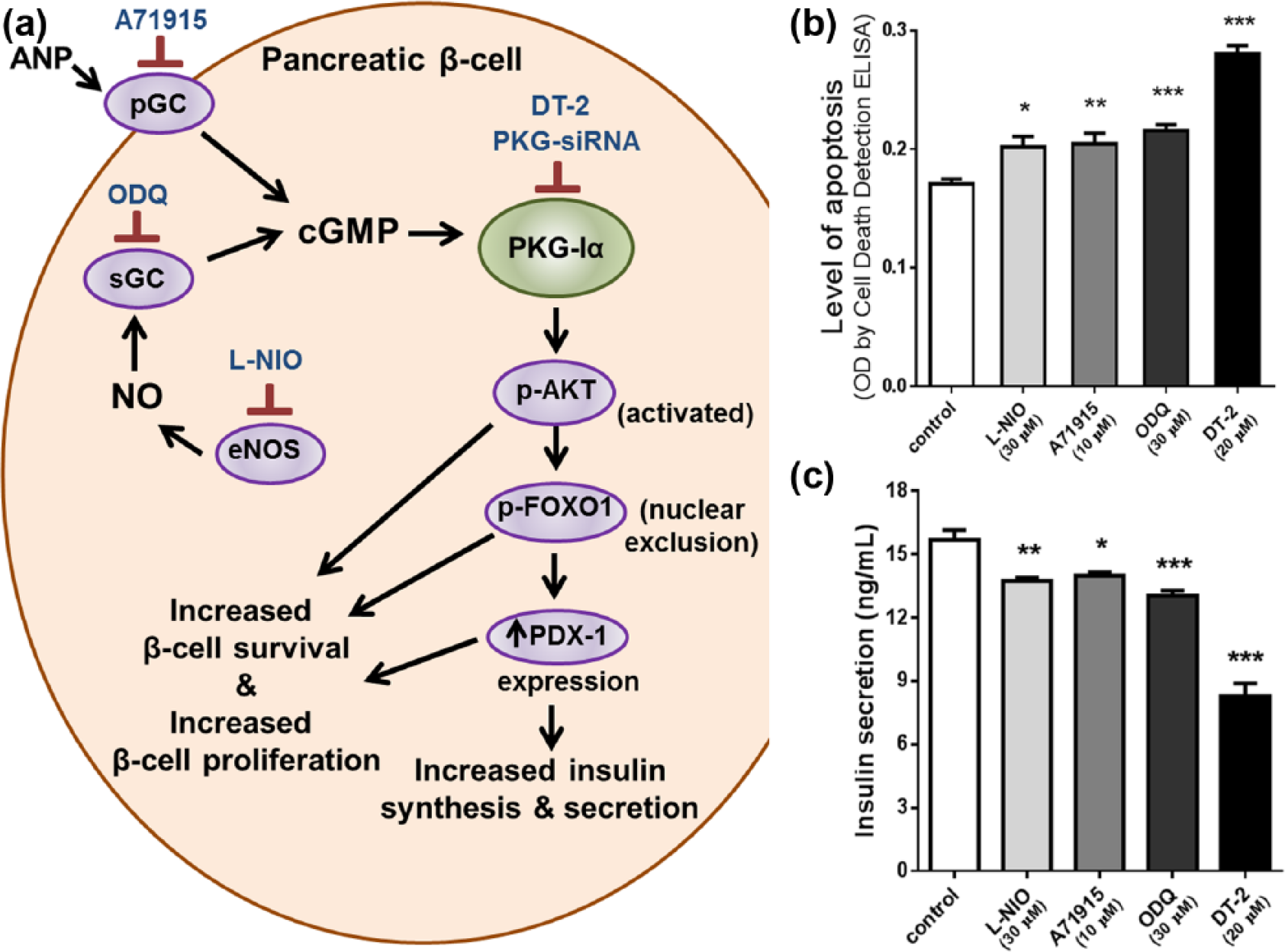
(a) Model of eNOS inhibitor L-NIO, ANP receptor blocker A71915, sGC inhibitor ODQ, PKG-Iα inhibitor DT-2 and siRNA gene knockdown of PKG-Iα suppressing Akt activation and downstream phosphorylation and nuclear exclusion of FOXO1, important for pancreatic β-cell phenotype, insulin secretion, survival and proliferation. (b) Induction of apoptotic cell death and (c) decreased insulin secretion in RINm5F β-cells were observed following inhibition of each of the key steps in NO/cGMP/PKG-Iα and ANP/cGMP/PKG-Iα signalling pathways. *p < 0.05, **p < 0.01, ***p < 0.001.
Inhibitors of NO/cGMP/PKG and ANP/cGMP/PKG pathways in RINm5F β-cells show an autocrine mechanism for endogenous NO and ANP in promoting cell survival and insulin secretion
In Figure 1(b), L-NIO, A71915, ODQ and DT-2 all increased apoptosis levels in RINm5F cells, suggesting an important role of endogenous NO-induced activation of sGC, endogenous ANP-induced activation of NPR-A/pGC and kinase/catalytic activity of PKG-Iα in protecting against spontaneous apoptosis. Figure 1(c) shows significant reduction in insulin secretion when each inhibitor was used. Especially noticeable is the large decrease in insulin secretion when PKG-Iα kinase activity was inhibited with DT-2.
Expression of PKG-I isoforms in mouse pancreatic islets and various pancreatic cell lines, and localization of PKG-I within insulin-containing β-cells of cell-dispersed mouse pancreatic islets
Figures 2(a) and 3(a) showed western blot analysis of protein expressions of PKG-Iα/β (primary antibody recognizing both PKG-I isoforms) in primary cultures of pancreatic islets from mice and pancreatic β-cell lines RINm5F, Beta-TC-6 (Figure 2(a)), INS-1, 1.1B4 (Figure 3(a)) and PANC-1 pancreatic ductal carcinoma cells (Figure 2(a)). We did not use PKG-Iα- and PKG-Iβ-specific antibodies because all of the commercially available antibodies that are claimed to be specific for PKG-Iα and PKG-Iβ were found by our laboratory to recognize multiple proteins other than PKG-Iα/β and thus could not be trusted. Western blots in Figures 2(a) and 3(a) show bands representing PKG-Iα/β in all cells, except (apparently) PANC-1 ductal carcinoma cells. From these data, we could not determine which isoform of PKG-I was expressed since both PKG-Iα and PKG-Iβ may have contributed to the apparent single band representing mouse pancreatic islets and all the pancreatic β-cell lines. Interestingly, samples of pancreatic islets often appear to give two closely migrating bands on western blots, as shown in Figure 2(a), suggesting expression of both PKG-I isoforms. To solve this problem, we then used NanoPro 1000 cIEF technology, which cleanly separates the two PKG-I isoforms, to determine relative expression levels of PKG-Iα and PKG-Iβ, as shown in our previous studies.7–9,34,40
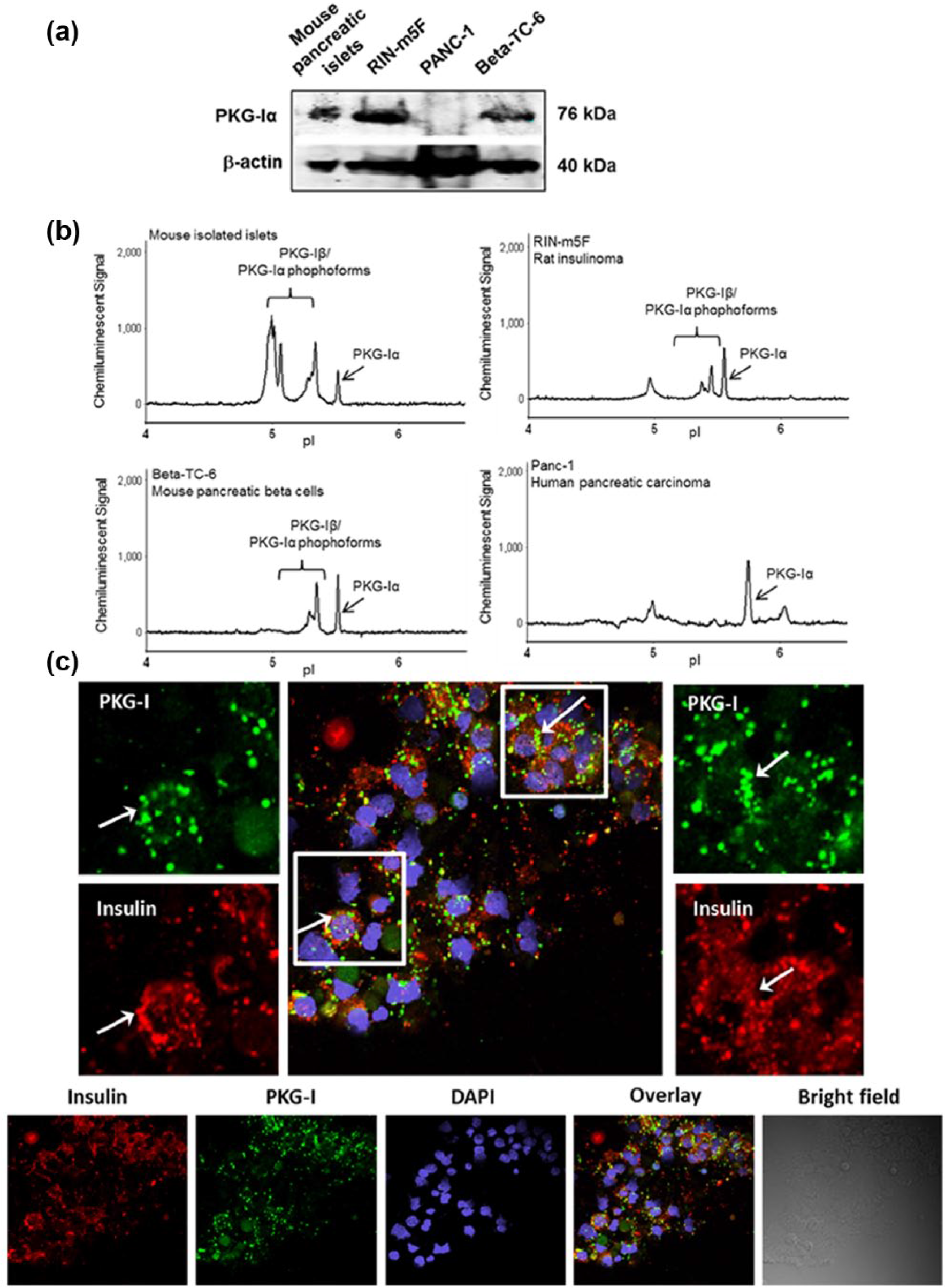
(a) Western blot analysis showing protein expressions of PKG-Iα in primary cultures of pancreatic islets from mice, two pancreatic β-cell lines (RINm5F and Beta-TC-6) and pancreatic ductal carcinoma PANC-1. (b) The ultrasensitive NanoPro 1000 system identified expressions of both PKG-Iα and PKG-Iβ isoforms in the same cells as in Figure 2(a). (c) Immunofluorescent imaging with confocal microscopy showing co-localization (yellow) of PKG-Iα (green) and insulin (red) in insulin-containing β-cells of mouse pancreatic islets.
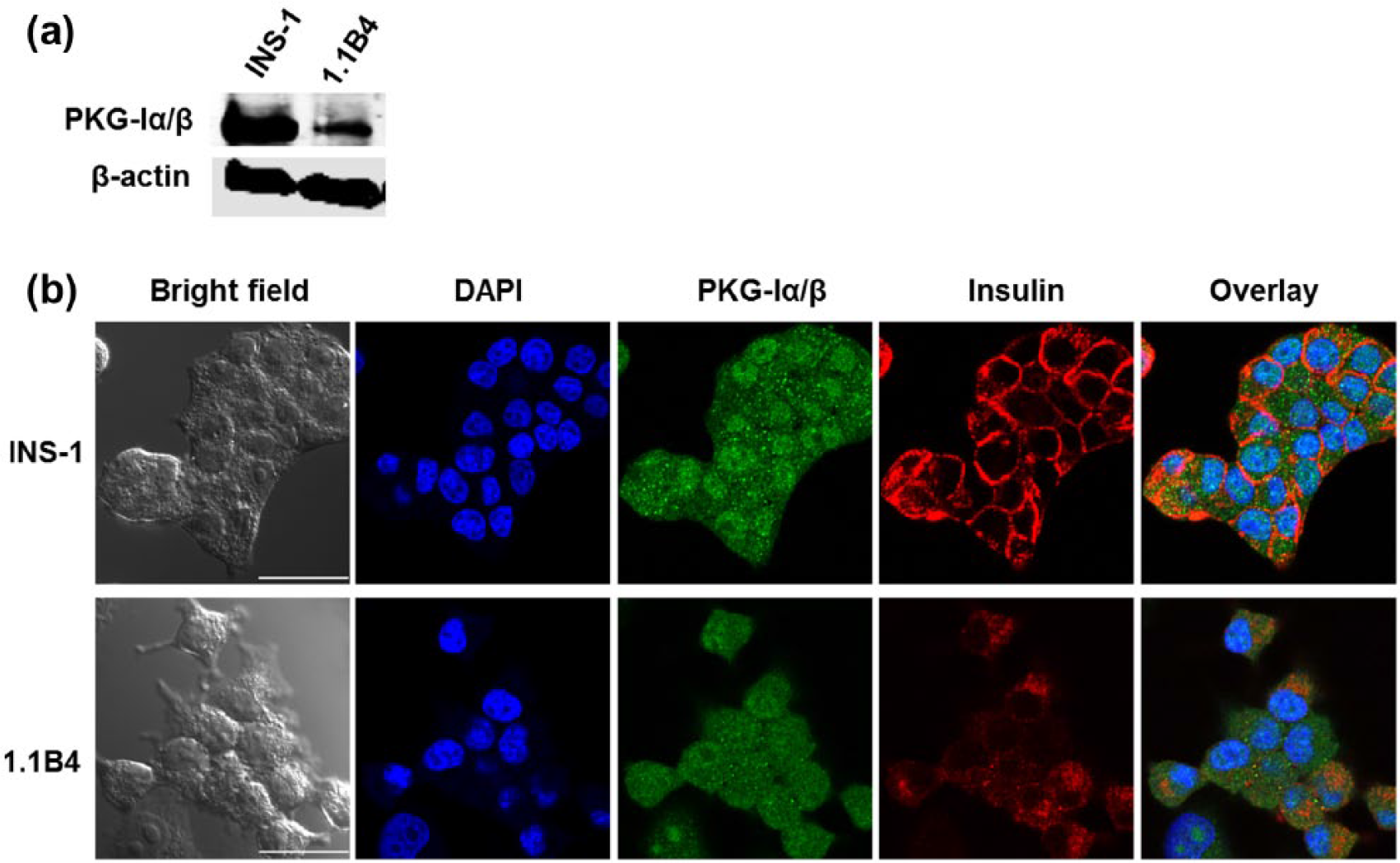
(a) Western blot analysis showing protein expressions of PKG-Iα in two pancreatic β-cell lines (INS-1 and 1.1B4). (b) Immunofluorescent imaging with confocal microscopy showing PKG-Iα (green) and insulin (red) expressions in INS-1 and 1.1B4 cells with some co-localization (orange or yellow). Scale bar = 25 µm.
Figure 2(b) shows cIEF analysis of the same samples as in Figure 2(a). The NanoPro 1000/cIEF electropherograms show that mouse pancreatic islets, RINm5F β-cells and Beta-TC-6 cells all express both isoforms of PKG-I. The pancreatic cancer cell line PANC-1 expressed predominately the PKG-Iα isoform, like reported previously for ovarian cancer cells.8,9
To study the localization of PKG-Iα/β and insulin, we performed immunofluorescent staining of mouse pancreatic islets, rat β-cell line INS-1 and the human β-cell line 1.1B4. Figure 2(c) shows expressions of PKG-Iα/β (green) and insulin (red) in samples of dispersed cells from mouse pancreatic islets. There was co-localization (yellow) of PKG-Iα/β and insulin in some of the cells, suggesting that PKG-Iα/β is expressed in insulin-containing β-cells. Interestingly, in the rat β-cell line INS-1 and the human β-cell line 1.1B4 (Figure 3(a)), PKG-Iα/β appears to be located especially in the nucleus, but also detected in the cytosol (punctated green spots) with some co-localization with insulin (red).
Gene knockdown of PKG-Iα expression, using siRNA specifically targeting PKG-Iα, increased spontaneous apoptosis, decreased DNA synthesis (BrdU incorporation) and decreased insulin secretion
To determine whether PKG-Iα plays a role in cell survival and proliferation, we performed functional assays using specific siRNAs to knockdown PKG-Iα in the pancreatic β-cell lines. Figure 4(a) shows increased levels of apoptosis, measured by Cell Death Detection ELISAPLUS, in RINm5F cells with the two PKG-Iα siRNAs (effectively verified previously in many other cell lines in our laboratory), versus control. More recently, we developed two other new PKG-Iα siRNA constructs, siPKG-Iα S1 and S2. As shown in the figure, PKG-Iα gene knockdown using siPKG-Iα S2 caused similar increase in apoptosis on all four β-cell lines, Beta-TC-6, RINm5F, INS-1 and 1.1B4 (Figure 4(d) to (g), respectively).
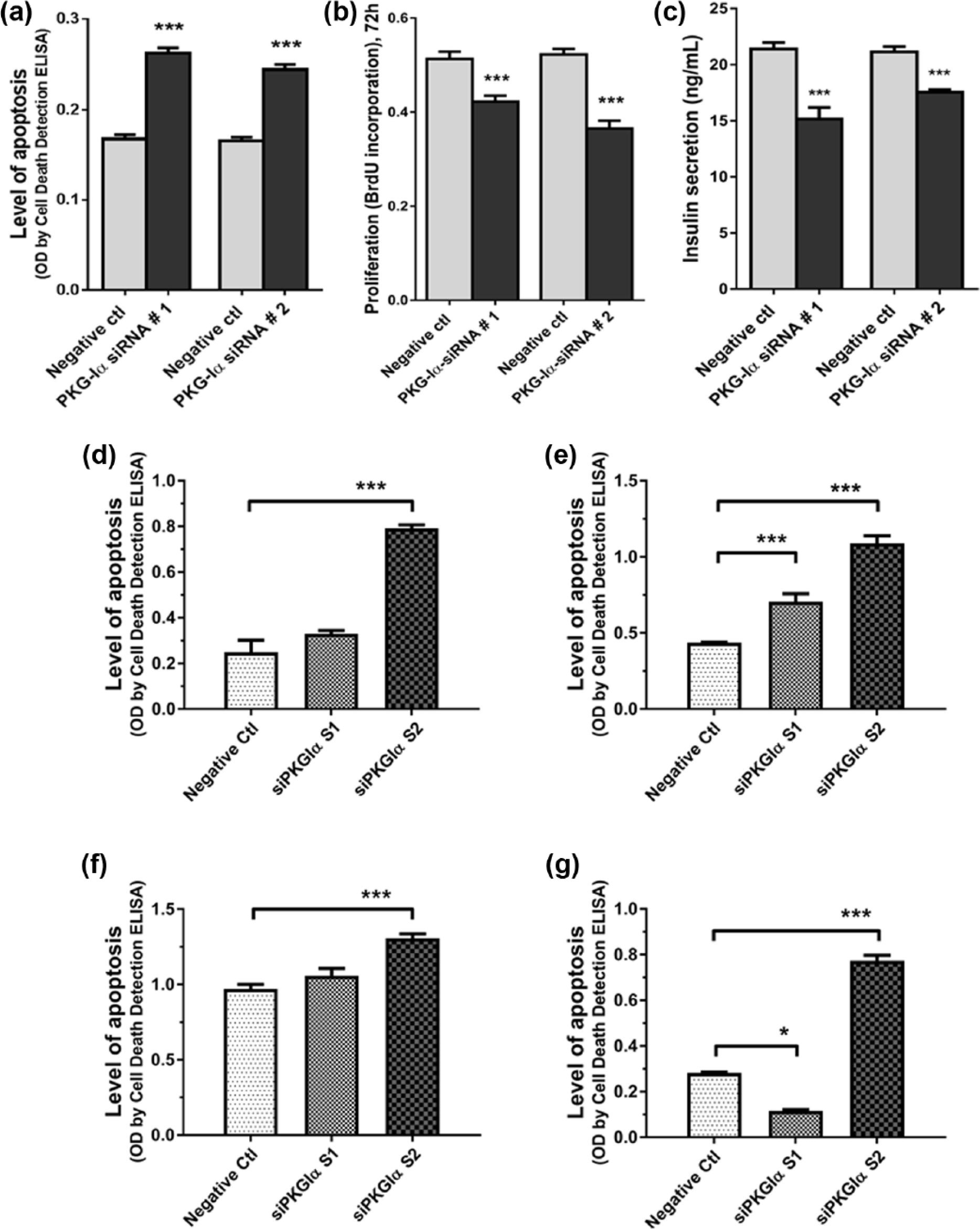
Specific PKG-Iα-siRNA knockdown of PKG-Iα expression using PKG-Iα siRNA 1 and 2 (a) increased apoptosis (Cell Death Detection ELISAPLUS), (b) decreased de novo DNA synthesis (BrdU incorporation) and (c) decreased insulin secretion in RINm5F β-cells. Specific PKG-Iα-siRNA knockdown of PKG-Iα expression using siPKG-Iα S1 and S2 also increased apoptosis (Cell Death Detection ELISAPLUS) in (d) Beta-TC-6, (e) RINm5F, (f) INS-1 and (g) 1.1B4 β-cells. *p < 0.05, ***p < 0.001.
Figure 4(b) shows decreased BrdU incorporation, index of de novo DNA synthesis, in RINm5F cells with PKG-Iα gene knockdown, versus control. Figure 4(c) shows significant decreases in insulin secretion in RINm5F cells with PKG-Iα gene knockdown, versus control.
Figure 5 shows confirmatory data using other techniques, that is, caspase-3/7 activity and MTT, to show that siRNA gene knockdown of PKG-Iα or inhibition of PKG-Iα kinase activity with DT-3 increases levels of apoptosis (higher caspase 3/7 activity) and decreases cell proliferation (lower MTT measurement) in RINm5F β-cells. These effects were confirmed in another pancreatic β-cell line, Beta-TC-6 cells (Figure 5(a) to (d)). PANC-1 pancreatic ductal carcinoma cells showed similar increased apoptosis and decreased proliferation when transfected with PKG-Iα-siRNA or exposed to PKG-Iα kinase activity inhibitor DT-3, showing survival/proliferation role of PKG-Iα in other pancreas-derived cells.
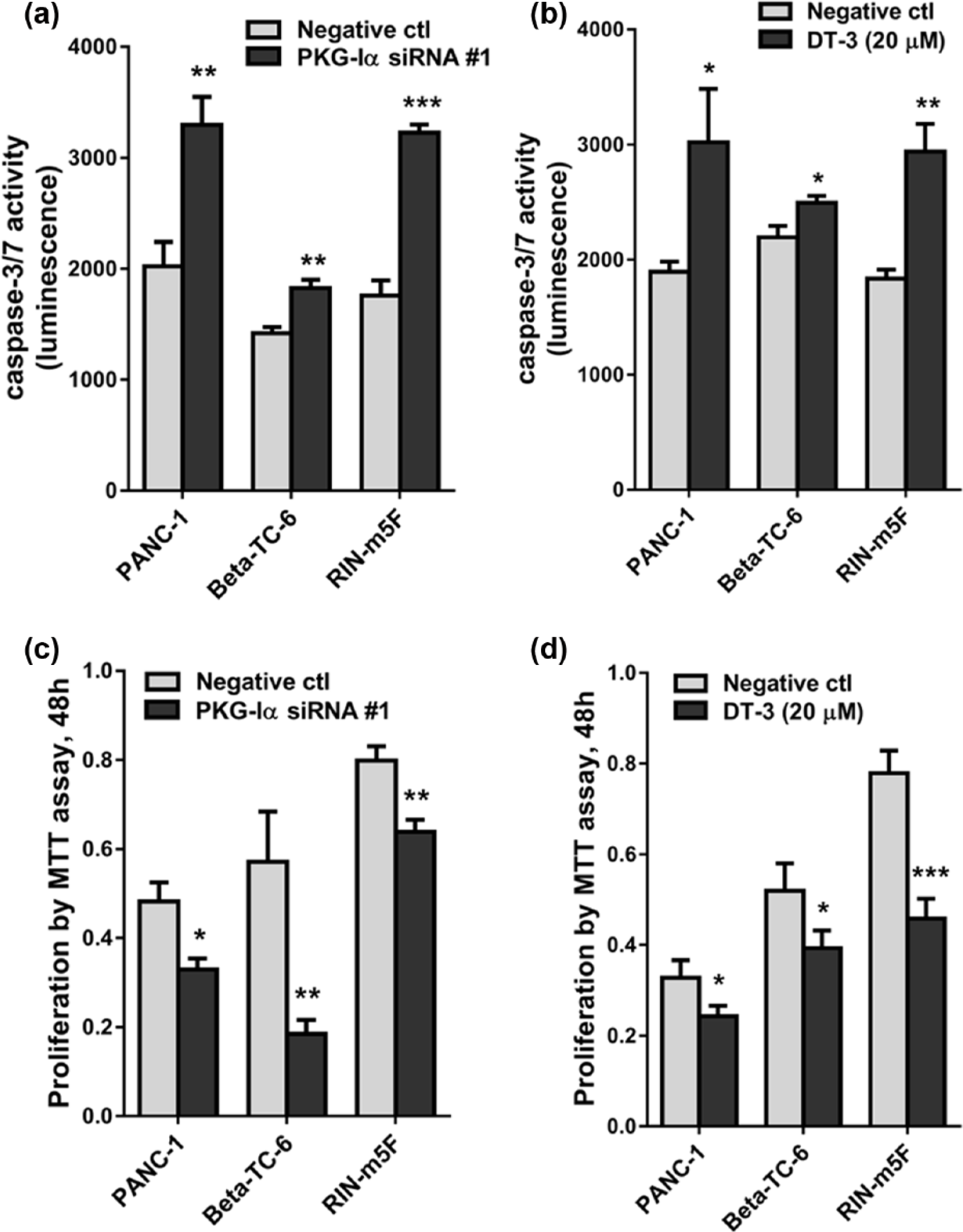
Specific PKG-Iα-siRNA knockdown and inhibition of PKG-Iα kinase activity with DT-3, with similar inhibitory effects as DT-2, 42 caused increase in (a, b) caspase-3/7 activity, a measure of apoptosis, and decreases in (c, d) proliferation by MTT assay, in RINm5F, Beta-TC-6 and PANC-1 cells. *p < 0.05, **p < 0.01, ***p < 0.001.
Data in Figure 6 further support the role of PKG-Iα in proliferation in four β-cell lines, Beta-TC-6 (panels (a) and (e)), RINm5F (panels (b) and (f)), INS-1 (panels (c) and (g)) and 1.1B4 (panels (d) and (h)), using DT-3 and the two other new PKG-Iα siRNA constructs, siPKG-Iα S1 and S2, measured by MTT (Figure 6(a) to (d)) and BrdU proliferation assay (Figure 6(e) to (h)). Treatment with DT-3 for 48 h caused a concentration-dependent decrease in proliferation in all four cell lines. Additionally, transfection with siPKG-Iα S2 decreased DNA synthesis in Beta-TC-6 and 1.1B4 cells. This is the first report showing the pro-proliferation role of PKG-Iα across many β-cell lines of different species (mouse, rat and human), as well as the first report about PKG-Iα in the human β-cell line 1.1B4.
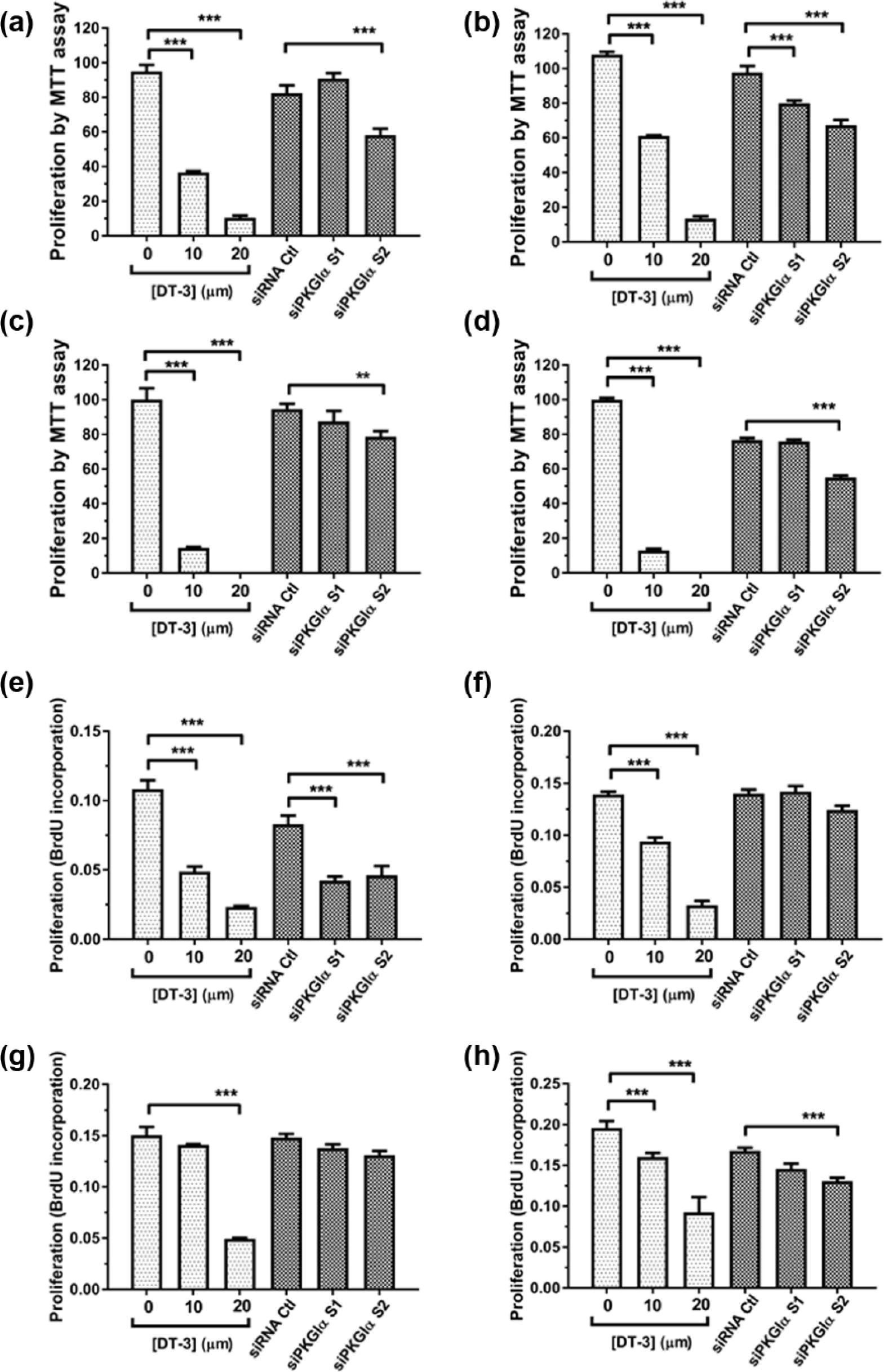
Specific PKG-Iα-siRNA knockdown of PKG-Iα expression using siPKG-Iα S1 and S2 and inhibition of PKG-Iα kinase activity with DT-3 decreased cell proliferation by MTT assay in (a) Beta-TC-6, (b) RINm5F, (c) INS-1 and (d) 1.1B4 cells and decreased de novo DNA synthesis (BrdU incorporation) in (e) Beta-TC-6, (f) RINm5F, (g) INS-1 and (h) 1.1B4 in the four β-cell lines. **p < 0.01, ***p < 0.001.
Suppression of AKT phosphorylation/activation and downstream phosphorylation of FOXO1 with concurrent decreases in PDX-1 expression in RINm5F β-cells with siRNA knockdown of PKG-Iα expression
Figure 7(a) and (b) shows that two different siRNA constructs targeting PKG-Iα, that is, PKG-Iα-siRNA 1 and 2, both successfully caused knockdown of PKG-Iα protein expression in RINm5F β-cells, as confirmed by western blot analysis, representing approximately 70% reduction in protein expression levels. To determine whether PKG kinase activity was also downregulated by the siRNA constructs, we assessed serine-239-phosphorylation of VASP, an actin-regulating protein often used to assess endogenous kinase activity of PKG-Iα.30,33,34,38 Figure 7(a) shows that not only protein levels of PKG-Iα are substantially reduced by PKG-Iα-siRNA constructs but also PKG-I kinase/catalytic activity.
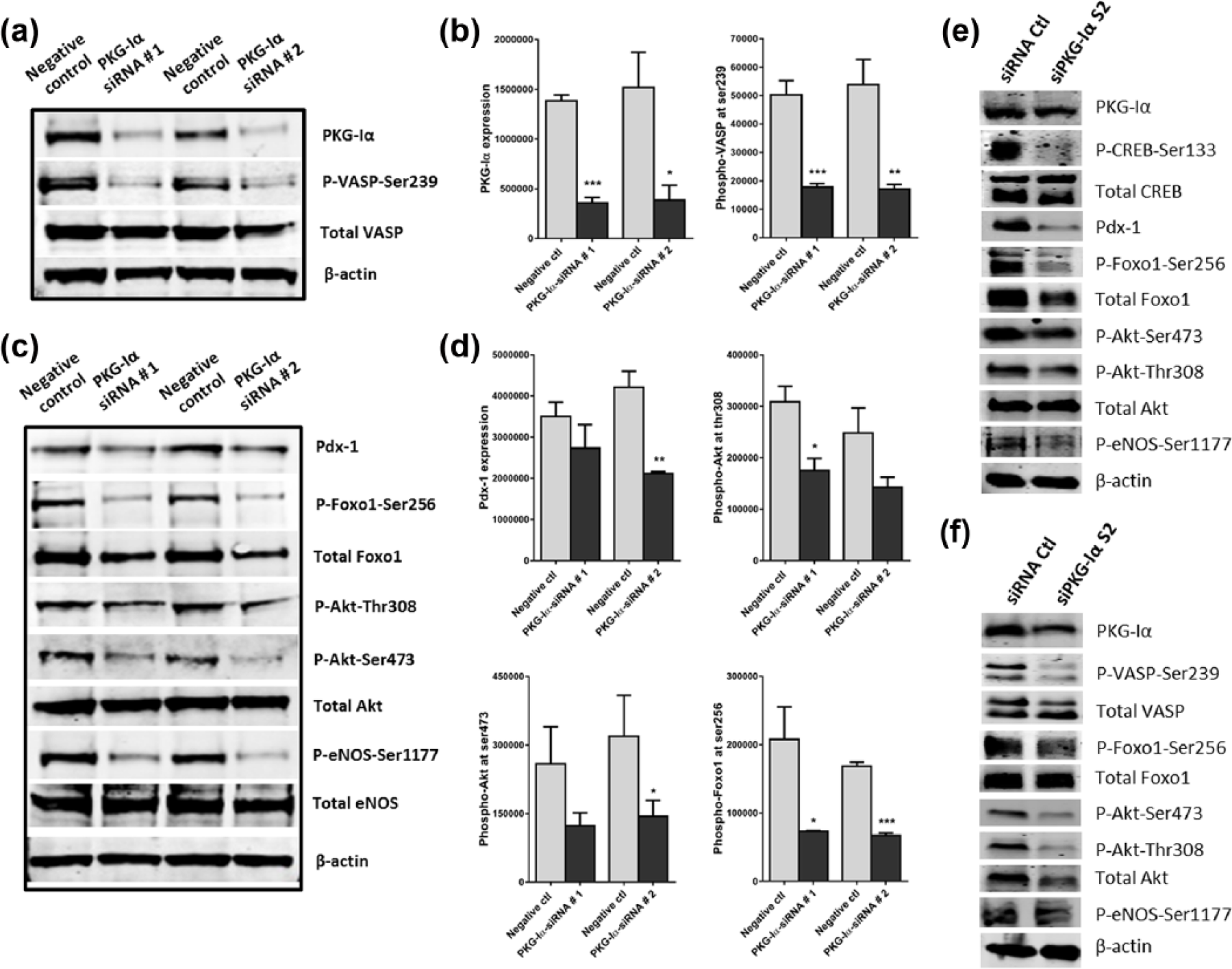
Western blot analysis of RINm5F β-cells showing (a) 70% reduction in PKG-Iα protein levels caused by knockdown using two different siRNA constructs, PKG-Iα-siRNA 1 and 2, and dramatic reduction in PKG-Iα kinase activity, measured by VASP serine-239-phosphorylation, (b) quantification of PKG-I expression and VASP phosphorylation, (c) effects on key components of the AKT/FOXO1/PDX-1 pathway and (d) quantification of the westerns in (c). Phosphorylation/activation of eNOS was also included as a downstream phosphorylation target of AKT, indicating AKT kinase activity. Western blot analysis showing the effects of siRNA-mediated knockdown of PKG-Iα expression using siPKG-Iα S2 on components of the AKT/FOXO1/PDX-1 pathway in (e) INS-1 and (f) 1.1B4 β-cells.
Figure 7(c) shows that in PKG-Iα-knockdown RINm5F β-cells, protein expression levels of PDX-1, a transcription factor necessary for β-cell development, survival and insulin secretion are also substantially reduced. Furthermore, phospho-AKT (serine-473 and threonine-308), indicative of activated AKT, was also decreased by PKG-Iα knockdown. Also, PKG-Iα knockdown caused reduced phospho-eNOS (phosphorylated by AKT at serine-1177) and phospho-FOXO1 (phosphorylated by AKT at serine-256). Because FOXO1 is a transcriptional repressor of PDX-1 expression, the increased FOXO1 nuclear localization caused by decreased phospho-FOXO1 would cause a suppression of PDX-1 expression, contributing to decreases in insulin secretion, cell survival and cell proliferation in pancreatic β-cells with damaged/reduced PKG-Iα kinase activity. Figure 7(e) and (f) shows confirmatory data on β-cell lines INS-1 and 1.1B4. PKG-Iα gene knockdown by siPKG-Iα S2 results in decreased PKG-Iα kinase activity (phospho-CREB at serine-133 in INS-1 and phospho-VASP at serine-239 in 1.1B4), PDX-1 expression, phospho-AKT (serine-473 and threonine-308) and phospho-eNOS (at serine-1177).
Co-culture of RINm5F β-cells with BMSCs/BM-MSCs, which secreted ANP, shows stimulation of β-cell proliferation
Our previous studies with the OP9/BMSCs showed that both NO and ANP serve as autocrine factors to activate PKG-Iα signalling pathway, promoting OP9/BMSC survival, proliferation and migration. 32 In this study, we determined the effects of co-culturing RINm5F β-cells with OP9/BMSCs, in a co-culture system (illustrated in Figure 8(a)) using Anopore inserts, which allowed only secretory factors to exchange in the media. Figure 8(b) shows that OP9/BMSC cells significantly stimulated cell proliferation of RINm5F β-cells, presumably via BMSC-secreted ANP activating PKG-Iα signalling pathway in RINm5F β-cells.
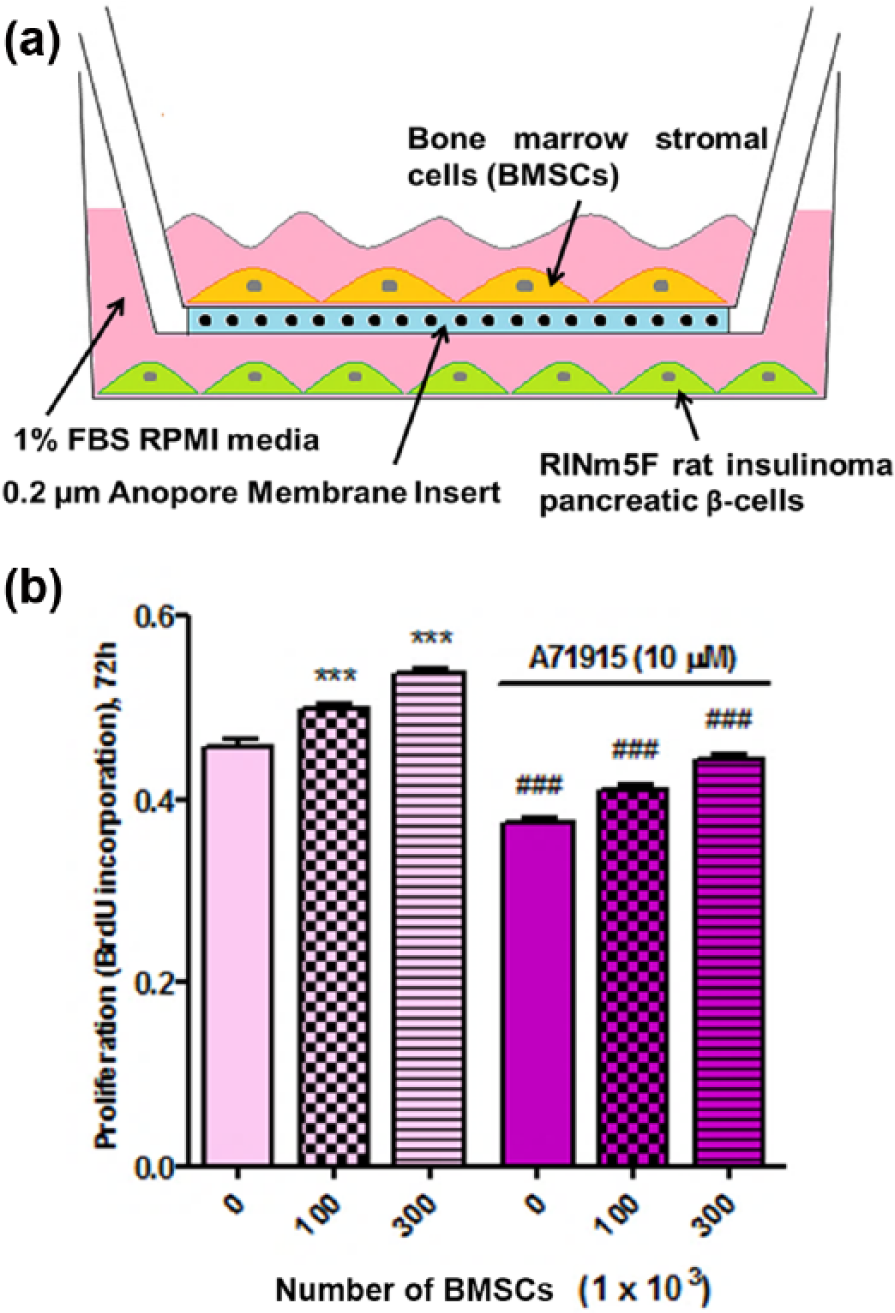
Bone marrow–derived stromal/mesenchymal stem cells (BMSCs/BM-MSCs, using OP9 cells), which synthesize and secrete ANP, were co-incubated with RINm5F β-cells to determine their effects on RINm5F cell proliferation (illustrated in (a)). (b) Incubation with 100,000 and 300,000 OP9/BMSCs caused significant increase in DNA synthesis/cell proliferation of RINm5F β-cells and this was blocked by the ANP receptor inhibitor A71915. ***p < 0.001, compared to no BMSC control. ###p < 0.001, compared to no A71915 groups.
We previously showed that A71915, a selective inhibitor of ANP receptor (NPR-1/pGC), blocked endogenous ANP/pGC/cGMP/PKG-Iα signalling autocrine loop and its stimulatory effects on cell survival and proliferation in OP9/(BMSCs/BM-MSCs). 32 In Figure 8(b), growth-stimulating effect of OP9/BMSCs on RINm5F β-cells was significantly reduced by A71915, indicating that at least part of the growth-promoting effects of BMSCs on RINm5F β-cells was due to secreted ANP.
Discussion
Data of this study may help explain the previous apparent conflict in the published data concerning the role of NO/cGMP/PKG signalling pathway in regulating apoptosis in pancreatic β-cells. Early data using HIT-T15 cells had shown that stimulation of the NO/cGMP/PKG signalling pathway with an NO donor increased levels of apoptosis, suggesting a pro-apoptotic role for NO/sGC/cGMP/PKG pathway. 11 In contrast, stimulation of carbon monoxide/sGC/cGMP/PKG pathway by exposure to exogenous carbon monoxide in another β-cell line, βTC3 cells, caused anti-apoptotic effects. 22 The opposite effects on apoptosis likely occurred because of very different levels of stimulation of the sGC/cGMP/PKG pathway, that is, high-level stimulation with the NO donor (used at pathological/toxicological concentrations) in the Loweth et al. study, likely causing activation of not only the PKG-Iα isoform but also a hyperactivation of the PKG-Iβ isoform, which can cause toxic effects within cells. The potential cellular mechanism of the cytotoxic effects of PKG-Iβ hyperactivation has been summarized in previous publications.7–9,20 In contrast, lower level stimulation of sGC/cGMP/PKG signalling caused by exposure to carbon monoxide, which protected β-cells against apoptotic cell death and improved glucose-lowering function of transplanted islets in diabetic animals, 22 likely involved a selective activation of the PKG-Iα isoform because of lower effects of carbon monoxide on activation of sGC. 24 The data of this study confirm the cytoprotective role of the PKG-Iα isoform, in which this cytoprotective kinase promotes activation of the AKT pathway, resulting in increased phosphorylation of FOXO1 and enhanced expression of PDX-1.
The human β-cell line 1.1B4 was generated by McCluskey et al. 43 by electrofusion of freshly isolated human pancreatic β-cells and the human PANC-1 epithelial cell line. They demonstrated that these cells preserved glucose sensitivity and produced insulin. These cells exhibit stable characteristics reminiscent of normal pancreatic β-cells, and it is therefore a suitable model for studying human pancreatic β-cell function. Furthermore, the 1.1B4 cell is a clinically relevant model because a very recent study showed that implantation of 1.1B4 cell pseudoislets significantly rescued diabetes and improved glucose tolerance. 44
Notably, this study is the first report to show that PKG-Iα protein expression is detectable in human β-cells and plays a role in cell survival and proliferation in the human β-cell line 1.1B4. From a translational perspective, data from this study provide useful insights that the cytoprotective and pro-proliferation roles of PKG-Iα are conserved in β-cells from rodent to human, and the human β-cell line 1.1B4 can be useful for screening therapeutic agents to treat both type-1 and type-2 diabetes.
Figure 9 illustrates a cellular model showing cytoprotective mechanisms of PKG-Iα, with the immediate downstream target proteins directly phosphorylated by PKG-Iα and their effects on cell survival and proliferation. In addition to PI3K/PDK1/AKT pathway being enhanced by PKG-Iα, shown in the present data and work by Doronzo et al., 23 the model also included other target proteins of PKG-Iα, such as Proto-oncogene tyrosine-protein-kinase Src (c-Src), Bcl-2-associated death promoter (BAD), CREB and VASP, shown by our laboratory to be directly phosphorylated/regulated by PKG-Iα.7–9,30–34,38,45 Also included is the contribution of other types of cells within the pancreatic islets, such as vascular endothelial cells that contribute physiological-level NO from eNOS, 39 M2 macrophages that contribute both carbon monoxide from its heme-oxygenase-1 39 and cytoprotective-level NO from low-level expression of iNOS and mesenchymal stem cells that contribute ANP (this study). Endothelial cells are known to release NO at levels that continually stimulate partial activation of PKG in nearby cells.17,29,46,47 The low/physiological-level NO, CO and ANP would all contribute to PKG-Iα activation and subsequent cytoprotection in pancreatic β-cells. During β-cell damage, as during type-1/type-2 diabetes and islet transplantation, all of these contributors would be expected to increase, helping to promote PKG-Iα kinase activity and its effects on promoting cell survival, cell proliferation and insulin secretion, to compensate for the damage of β-cells.
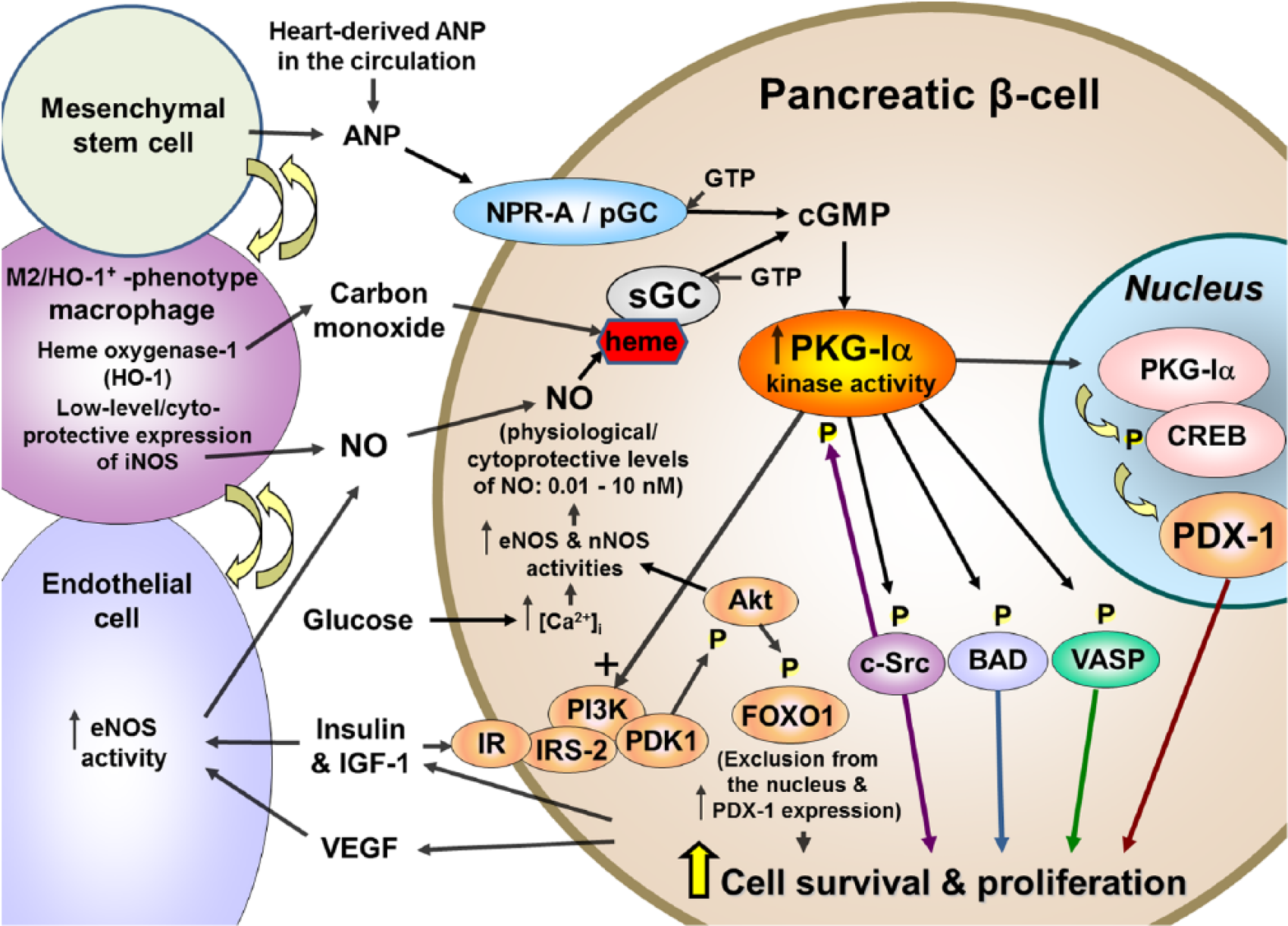
Model illustrating the proposed cellular mechanisms involving PKG-Iα in promoting cell survival and proliferation in pancreatic β-cells.
Genetic overexpression of PKG-Iα in BM-MSCs dramatically improves survival of these cells and their ability to stimulate cardiac muscle regeneration when these cells are used in stem-cell-based therapy for treating myocardial infarction. 48 Similar regenerative effects would be expected if PKG-Iα is kept active and functional in pancreatic β-cells during stressful conditions like type-1/type-2 diabetes and transplantation therapy.
In summary, the present data show cytoprotective effects of PKG-Iα kinase activity and downstream regulation of AKT/FOXO1 signalling pathway in four pancreatic β-cell lines, murine Beta-TC-6, rat RINm5F, rat INS-1 and human 1.1B4, defined using both pharmacological inhibitors and siRNA/genetic knockdown. The data illustrate the key role played by PKG-Iα in promoting β-cell survival and proliferation. Co-culturing with BMSCs/BM-MSCs, which synthesize and release ANP, was shown to promote RINm5F β-cell proliferation, suggesting a potential role of these cells, via their secretion of ANP and downstream activation of PKG-Iα in β-cells, as potential therapy in promoting β-cell regeneration. Overall, the present data have therapeutic implications for development of new therapeutic agents for treatment of both type-1 and type-2 diabetes as well as for improving β-cell survival/function in isolated pancreatic islets used for transplantation therapy.
Footnotes
Acknowledgements
The authors thank Dr Renee Coffman and Dr Harry Rosenberg, co-founders of Roseman University of Health Sciences, for their ideas in diabetes research that contributed to the early stages of this project and for their support of the specialized equipment needed for completing this project. They also thank Mary G Johlfs, Director of Research Operations at Roseman’s Summerlin Campus, for her help in overseeing the specialized instrumentation (e.g. NanoPro 1000 capillary isoelectric focusing instrument and confocal microscope) used in this project. J.C.W. and V.V. are co-first authors.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
This work was supported by Internal Funding from Roseman University of Health Sciences and a grant from the US Department of Defense (Grant no. W81XWH-07-1-0543; R.R.F.).