
Abstract
Type 2 diabetes mellitus escalates the risk of heart failure partly via its ability to induce a cardiomyopathic state that is independent of coronary artery disease and hypertension. Although the pathogenesis of diabetic cardiomyopathy has yet to be fully elucidated, aberrations in cardiac substrate metabolism and energetics are thought to be key drivers. These aberrations include excessive fatty acid utilisation and storage, suppressed glucose oxidation and impaired mitochondrial oxidative phosphorylation. An appreciation of how these abnormalities arise and synergise to promote adverse cardiac remodelling is critical to their effective amelioration. This review focuses on disturbances in myocardial fuel (fatty acids and glucose) flux and energetics in type 2 diabetes, how these disturbances relate to the development of diabetic cardiomyopathy and the potential therapeutic agents that could be used to correct them.
Keywords
Introduction
Type 2 diabetes mellitus (T2DM) is a global pandemic that is associated with a more than 2-fold greater risk of developing heart failure (HF), 1 and a 60%–80% greater probability of death in those with established HF.2,3 While accelerated coronary artery disease and hypertension largely explain these statistics, 4 evidence suggests that T2DM itself can drive adverse cardiac remodelling and give rise to a diabetic cardiomyopathy (DiCM).5–7 Although hotly debated, DiCM is reportedly evident in up to 60% of patients with T2DM and is characterised by unexplained myocardial hypertrophy and fibrosis, with left ventricular (LV) diastolic (±systolic) impairment.8–11 At the cardiomyocyte level, defects in excitation–contraction coupling, calcium mishandling and increased oxidative stress are present.12–14 More importantly, profound alterations in myocardial substrate metabolism and energetics have been shown.15,16 Crucially, these metabolic derangements not only precede cardiac structural and functional changes, but their early correction in animal models of T2DM aborted the development of DiCM.15,17 Consequently, metabolic abnormalities of the heart are promising therapeutic targets whose amelioration might improve outcomes in T2DM.
Here, after recounting normal myocardial metabolism and energetics, we review how these processes are disturbed in T2DM, how disturbances relate to the genesis of DiCM and the potential therapeutic agents that could be used to ameliorate them.
Normal myocardial metabolism
Cardiac energetics
The heart is an astonishing organ. Over an average human lifespan, it beats ~2–3 billion times and circulates ~200 million litres of blood. To sustain such a high workload, the heart can consume up to 30 kg of adenosine triphosphate (ATP) daily (~75–100 times its own weight) which is equivalent to the energy needed to climb a 100-storey building. 18 Thus, the heart is the highest energy consuming organ of the body, and subtle energetic deficits can rapidly induce contractile dysfunction.19,20 Uninterrupted ATP generation is dependent on the continuous supply of oxygen and fuel substrates and on the integrity of oxidative phosphorylation (OxPhos) which produces virtually all the hearts’ ATP.21,22 Of the generated ATP, ~60%–70% fuels cardiac contractions with the remainder used for ion pumps such as the sarcoplasmic reticulum Ca2+-ATPase (SERCA). While the heart can switch its substrate [fatty acids (FAs), glucose, ketones, lactate, amino acids] preference depending on workload, oxygen supply and hormones, its two main energetic substrates are FAs and glucose. 22
FA uptake and utilisation
FAs are the preferred fuel substrates and account for 70%–90% of myocardial ATP generation (Figure 1). 23 They circulate in plasma as triglyceride (TG)-rich lipoproteins and, to a lesser extent, bound to albumin. Endothelial lipoprotein lipases cleave lipoproteins to release free FAs which then enter cardiomyocytes via specific transporters. 24 Once in the cytosol, free FAs are esterified to fatty acyl-CoA which is either stored in the intracellular TG pool (10%–30%) or transported into the mitochondrion for β-oxidation (70%–90%). 25 Carnitine palmitoyltransferase (CPT)-I is the rate-limiting enzyme of FA metabolism. It translocates fatty acyl-CoA into mitochondria by converting it to fatty acyl-carnitine. Once in the mitochondrion, fatty acyl-carnitine is converted back to fatty acyl-CoA by CPT II for β-oxidation. 24 Overall, FA metabolism is principally regulated by the transcription factor peroxisome proliferator-activated receptor-α (PPAR-α) which, upon activation by high myocardial FA levels, upregulates FA uptake and utilisation and suppresses glucose oxidation. 26 In turn, increased glucose oxidation can reduce FA utilisation. This reciprocal inhibition is termed the ‘Randle cycle’. 27 Although FAs generate more ATP than glucose for each molecule metabolised, the ATP production to oxygen consumption ratio is lower for FAs (2.33 vs 2.58). Thus, increased FA oxidation reduces cardiac efficiency (cardiac work/myocardial oxygen consumption).24,28
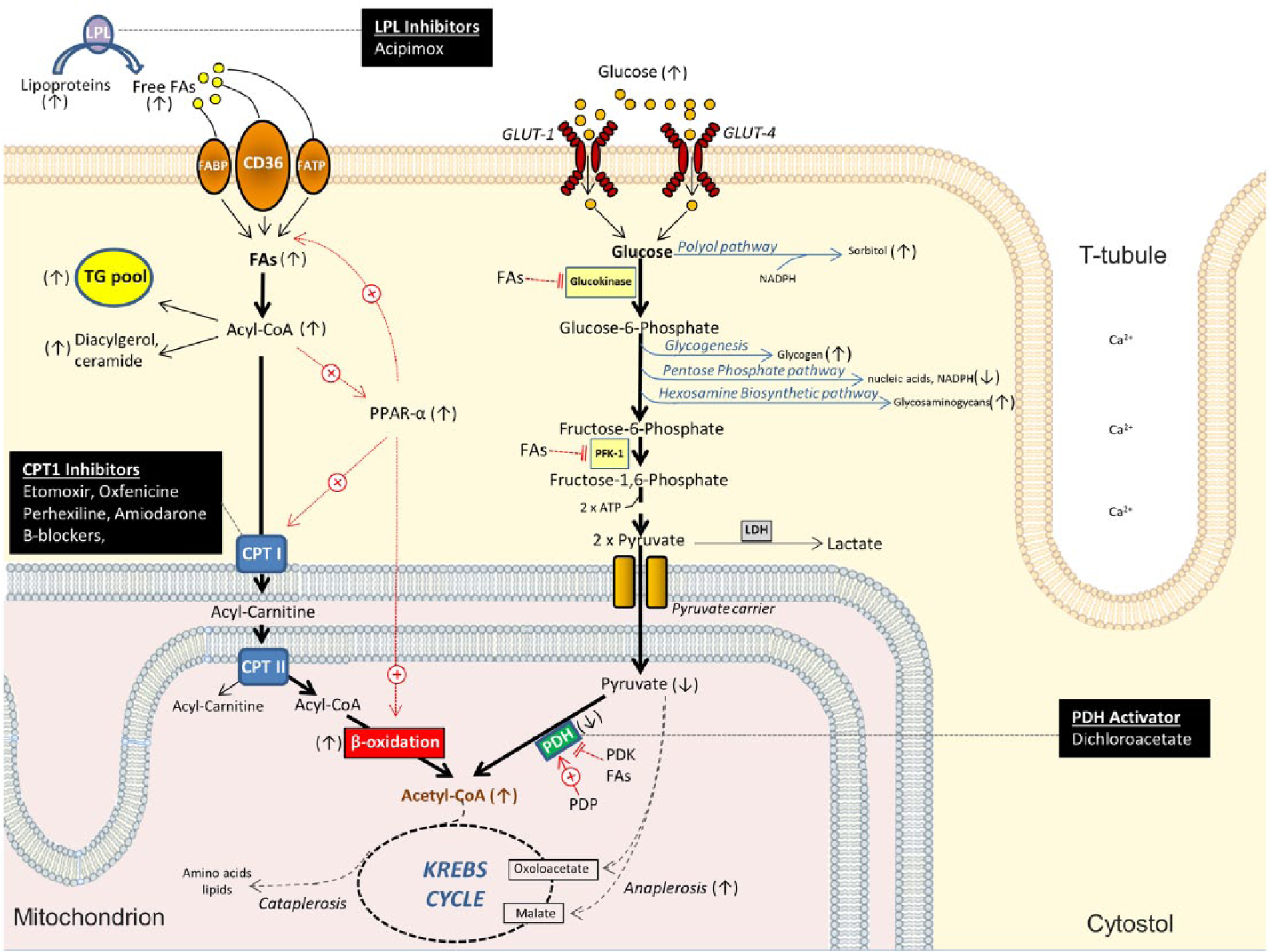
Fatty acid and glucose metabolism in type 2 diabetic cardiomyocytes. Substrate uptake occurs via specific transporters. Fatty acids (FAs) principally undergo β-oxidation in mitochondria under the control of peroxisome proliferator-activated receptor (PPAR)-α. Glucose molecules flux through diverse metabolic fates but mainly undergo glycolysis (pathway in bold). Arrows in parentheses denote the direction of change in type II diabetic cardiomyocytes compared to normal cells. Potential therapeutic agents are listed in the black boxes.
Glucose uptake and utilisation
Glucose oxidation accounts for 10%–30% of cardiac ATP production. It is initiated by glucose entry into cardiomyocytes via glucose transporter (GLUT)-1 and -4 (Figure 1). GLUT-1 mediates basal insulin-independent glucose uptake, while GLUT-4 is insulin responsive. 29 Once in the cell, limited amounts of glucose (<5%) enter the polyol (or sorbitol-aldose reductase) pathway which consumes reducing equivalents [nicotinamide adenine dinucleotide phosphate (NADPH)] to generate sorbitol. 30 However, the vast majority of imported glucose is converted to glucose-6-phosphate by glucokinase. Glucose-6-phosphate is then channelled into glycolysis (~85%), glycogenesis (<5%), the pentose phosphate pathway (PPP; <5%) for nucleic acid and NADPH synthesis 31 or the hexosamine biosynthetic pathway (HBP; <5%) for the production of precursors of glycosaminoglycans. 32 During glycolysis, phosphofructokinase (PFK)-1 catalyses the rate-limiting step of early glycolytic reactions that generate pyruvate which is then oxidatively decarboxylated by pyruvate dehydrogenase (PDH) to form acetyl-CoA. PDH is a critical site of regulation. It is inhibited by pyruvate dehydrogenase kinase (PDK) and stimulated by pyruvate dehydrogenase phosphatase (PDP). In turn, PDK is stimulated by PPAR-α, FAs, acetyl-CoA and nicotinamide adenine dinucleotide (NADH), while PDP is stimulated by elevated intracellular Ca2+ levels.33,34 Under anaerobic conditions, pyruvate is converted to lactate or shuttled into anaplerotic pathways (Figure 1).
Krebs cycle and mitochondrial OxPhos
Irrespective of substrate preference, the final end-product of fuel oxidation is acetyl-CoA which enters the Krebs cycle to generate reducing equivalents [NADH and flavin adenine dinucleotide (FADH2)] for OxPhos which produces >95% of myocardial ATP (Figure 2). 35 Because Krebs intermediates are continuously siphoned off for amino acid, lipid and nucleic acid biosynthesis (cataplerotic processes), uninterrupted NADH and FADH2 production depends on anapleurosis, which is the replenishment of Krebs moieties independently of acetyl-CoA. 36 Once formed, NADH and FADH2 release electrons to the electron transport chain (ETC), inducing sequential redox reactions that generate the proton gradient that drives ATP synthesis (Figure 2). Phosphocreatinine (PCr) then links ATP production to ATP consumption. Even under normal conditions, OxPhos is not fully efficient as 1%–2% of electrons leak from the ETC to form reactive oxygen species (ROS), and uncoupling proteins (UCPs) partially dissipate the proton gradient to generate heat. 37 Besides Krebs cycle flux, molecular oxygen, ETC proteins and coenzyme Q10, OxPhos is also dependent on iron. This is because iron is a component of haem within ETC and Krebs cycle enzymes and of iron–sulfur clusters that execute the electron transfer function of most ETC complexes.38,39 Consistent with its high energy demands, the myocardium has the highest mitochondrial density (35% of cardiomyocyte volume versus 3%–8% in skeletal and smooth muscle cells) of any organ, and its mitochondria exhibit the greatest number of cristae which enhance OxPhos.40–42
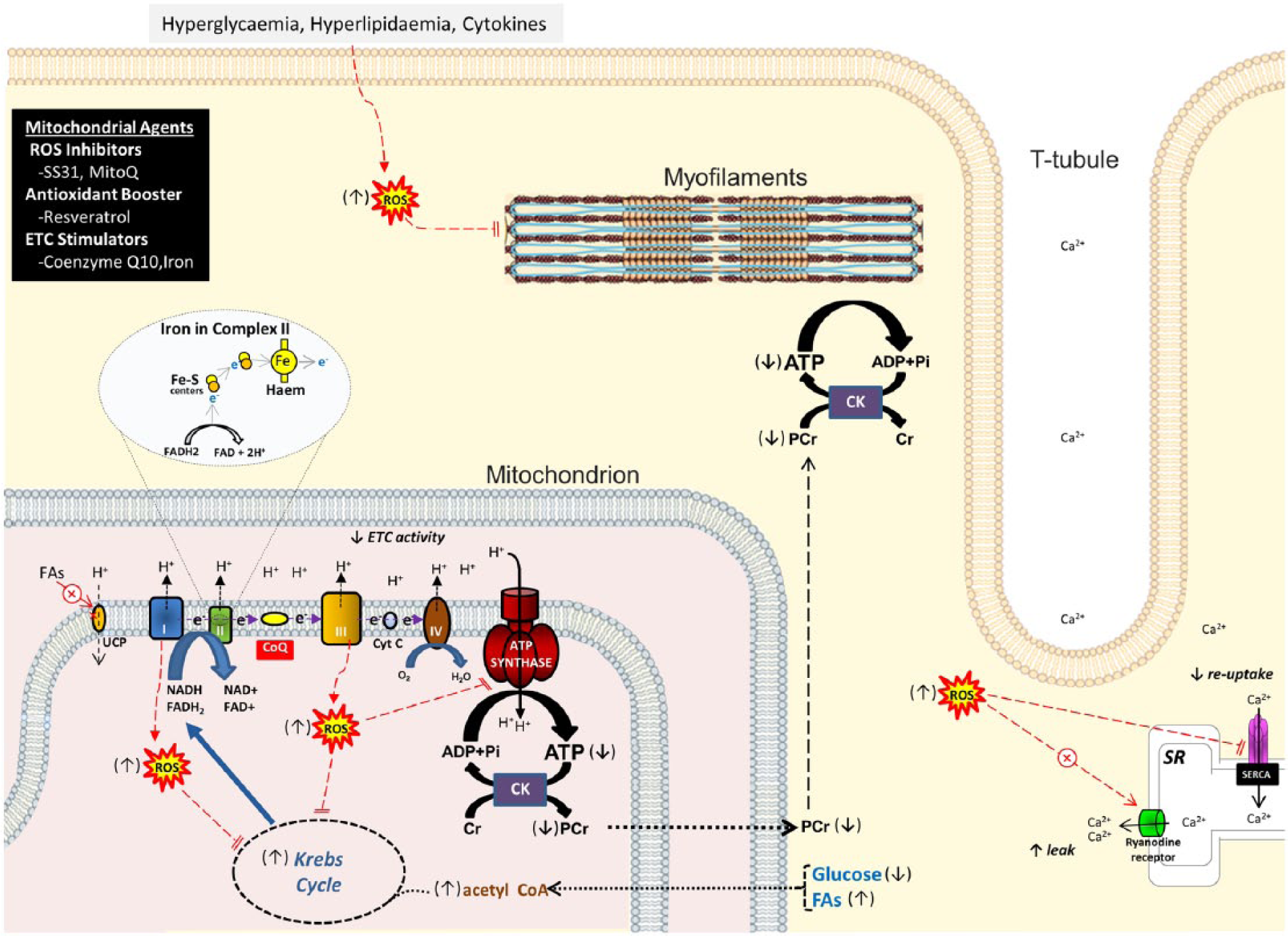
Mitochondrial oxidative phosphorylation in type 2 diabetic cardiomyocytes. Substrate oxidation generates reducing equivalents via the Krebs cycle which feed electrons (e−) into the ETC. Electron flow drives a proton gradient which powers ATP synthesis. Mitochondrial and cytostolic ROS impair oxidative phophosphorylation, myofilament function and calcium homeostasis. Iron mediates electron transfer in many ETC complexes. Arrows in parentheses denote the direction of change in type II diabetic cardiomyocytes compared to normal cells. Potential mitochondrial therapeutic agents are listed in the black box.
Myocardial metabolism in T2DM
Abnormal cardiac FA uptake and utilisation in T2DM
The hearts’ preference for FAs as energetic substrates is exaggerated in T2DM. This is due to the increase in plasma FA levels as a result of increased lipolysis from insufficient insulin action, increased hepatic TG production and inefficient adipocyte TG storage. The enhanced delivery of FAs to the sarcolemma then elevates myocardial FA uptake and TG stores. 43 As a result, myocardial FA abundance activates PPAR-α which amplifies the reliance on FAs further by upregulating FA uptake, storage and β-oxidation, while suppressing glucose utilisation. 26 Increased PPAR-α activity is evident in rodent models of DiCM, and hearts from PPAR-α knock-out mice do not remodel when rendered insulin resistant. 44 In patients with T2DM, a twofold increase in cardiac FA oxidation has been described, 45 and aberrant FA metabolism has been correlated to adverse cardiac changes (Figure 3).
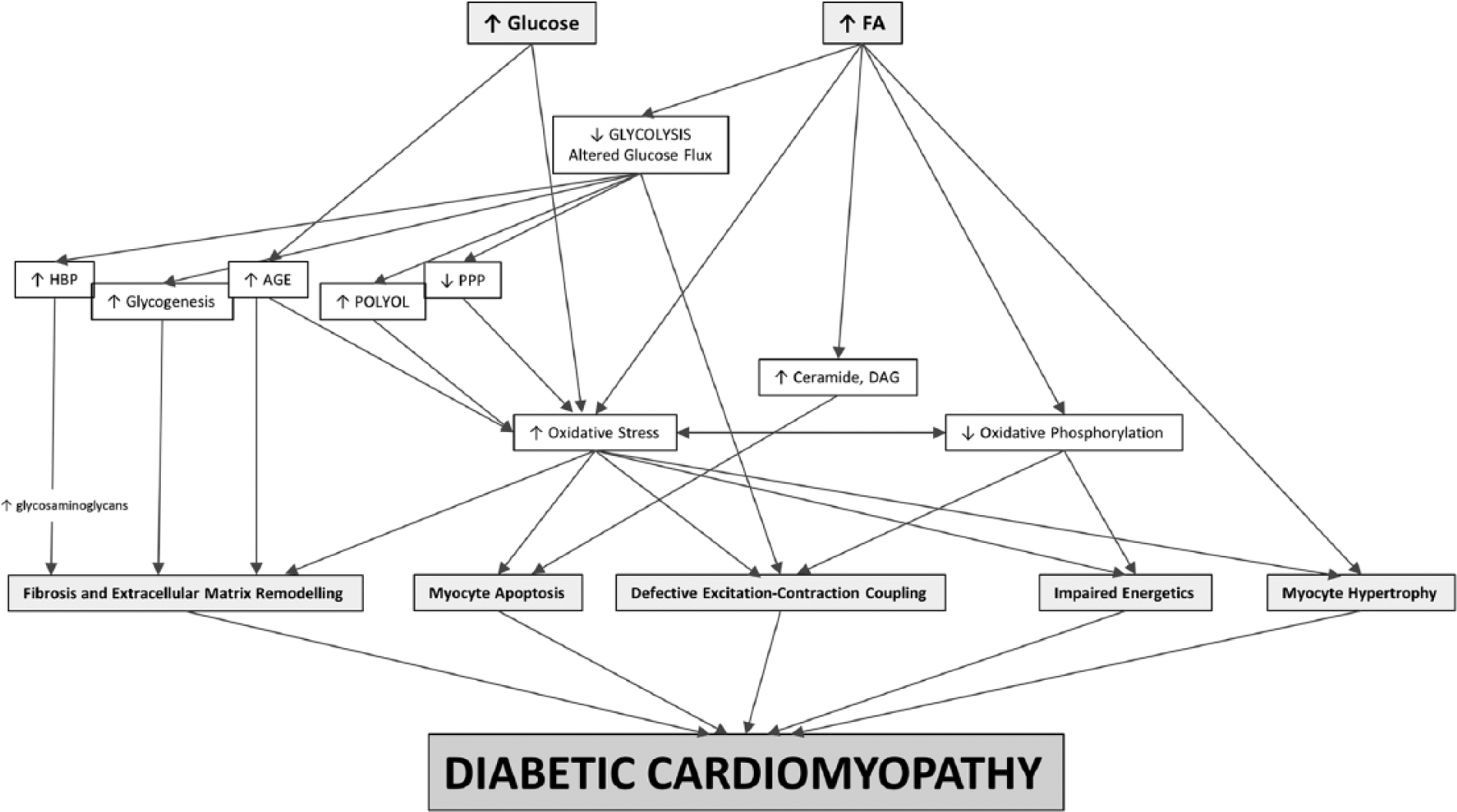
Changes in metabolism and their relevance to diabetic cardiomyopathy. Fatty acid (FA) abundance impairs glycolysis, shunts glucose towards more pathological pathways and leads to other abnormalities that plausibly drive diabetic cardiomyopathy. Hyperglycaemia augments levels of oxidative stress and advanced glycation end-products (AGE).
Excessive FA storage is thought to be cytotoxic and myocardial steatosis is associated with cardiac dysfunction in T2DM. Up to twofold increases in myocardial TG content have been shown in patients with T2DM, with higher TG levels related to lower LV diastolic and systolic function.46–48 FA accumulation within the heart is postulated to increase mitochondrial ROS by augmenting β-oxidation and ETC activity. 49 Elevated ROS levels are, in turn, implicated in myocardial hypertrophy, defective excitation–contraction coupling and cardiac inefficiency via mitochondrial uncoupling (Figure 2).28,50 Additionally, increased lipid intermediary metabolites such as ceramide and diacylglycerol can derange signalling and can trigger cardiomyocyte apoptosis directly or via cytochrome c release. 51 These events underlie ‘lipotoxicity’ which is implicated in the genesis of DiCM. 52 Because of its higher oxygen cost (~86% greater myocardial oxygen consumption compared to glucose), increased FA metabolism further diminishes cardiac mechanoenergetic efficiency. 53
Abnormal cardiac glucose uptake and utilisation in T2DM
Despite systemic hyperglycaemia, and appropriate glucose uptake in some studies, cardiac glucose oxidation is reduced by 30%–40% in patients with T2DM.45,54–56 This is largely attributed to FA excess in the myocardium. FAs reduce pyruvate synthesis by inhibiting glucokinase and PFK-1 and attenuate pyruvate oxidation by inhibiting PDH directly, and indirectly via augmented PPAR-α, acetyl-CoA and NADH levels.57–60 Reductions in glycolysis are accompanied by decreases in PPP flux and increases in polyol, HBP and glycogeneic pathways that may have pathological relevance.61–64
Hyperglycaemia and the end-products of abnormal glucose disposition possibly mediate some of the features of DiCM. Hyperglycaemia elevates cardiomyocyte glucose levels which can non-enzymatically glycate proteins to form advanced glycation end-products (AGEs). AGEs enhance ROS production and can cross-link and damage macromolecules such as collagen (leading to myocardial fibrosis and stiffness), SERCA (leading to diastolic impairment) and the ryanodine receptor (leading to contractile impairment).65–67 Increased polyol flux and decreased PPP activity reduce NADPH levels which increases oxidative stress.61,62 Augmented HBP flux increases the production of glycosaminoglycans, which can contribute to matrix remodelling, myocardial fibrosis and impaired SERCA activity. 63
Cardiac mitochondrial OxPhos defects in T2DM
Cardiac mitochondria exhibit diminished rates of OxPhos and a greater uncoupling of respiration from ATP generation in T2DM. This occurs despite evidence of increased cellular anaplerosis 68 and an increased delivery of Krebs cycle end-products (NADH and FADH2) to the ETC. This implies that factors downstream of the Krebs cycle limit OxPhos. Indeed, in the hearts of animals and humans with T2DM, a consistent reduction in the amount and catalytic activity of ETC complexes have been shown, and ETC dysfunction has been linked to oxidative stress.69–73 Reductions in OxPhos also result from FA-mediated upregulation of UCPs which uncouple mitochondria, and to attenuations in mitochondrial density (only in animal models), size and internal complexity.14,74–77 In patients with T2DM, reductions in mitochondrial size relate to impaired mitochondrial fusion with decreased mitofusin-2 expression. 75 In rodent models, lower PCr replenishment by creatine kinase has also been shown, 78 suggesting that defects in energy transduction might accompany those in energy generation.
Mitochondrial dysfunction is associated with abnormal myocardial structure and function in T2DM. Reductions in OxPhos manifest in decreases in myocardial PCr and ATP content which have been shown in animals and humans with T2DM. Greater energetic deficits, as reflected by lower cardiac PCr/ATP ratios, related to lower ETC activities, lower exercise capacity and poorer diastolic function.16,79,80 Moreover, in atrial trabeculae excised from surgical patients with T2DM, reductions in OxPhos were associated with impaired contractility. 71 While mitochondrial dysfunction could be deleterious solely via its ability to limit ATP supply to myofibrils (leading to systolic impairment) and SERCA (leading to diastolic impairment), it also augments ROS production which is implicated in cardiac remodelling.72,73 Increased ROS production results from the abundance of electrons delivered to the ETC (due to increased FA oxidation) which, because the ETC is defective, increasing leak to form ROS. Although mitochondrial dysfunction, lipotoxicity and glucotoxicity plausibly contribute to DiCM, it is unclear whether they are markers or mediators of cardiac remodelling. This uncertainty reflects the paucity of trials targeting cardiac metabolism in patients with T2DM.
Therapeutic targeting of cardiac metabolism in T2DM
Diabetic and HF medications
As unexplained cardiomyopathy is possibly driven by T2DM itself, good diabetic control seems intuitively important for preventing DiCM. However, only animal studies have shown that achieving early euglycaemia arrests the development of DiCM, and that certain diabetic drugs can have specific anti-remodelling effects.15,17 For example, in ex vivo mice hearts, glucose–insulin infusions improved glucose oxidation and contractile efficiency, while incretin-based therapies (Liraglutide and Exendin-4) reversed steatosis, oxidative stress and SERCA downregulation.81–84 Limited data exist in humans, and some of them are at odds with findings in animals. In a retrospective analysis, biguanide (metformin) use was associated with reduced natiuretic peptide levels and lower cardiovascular morbidity and mortality. 85 In contrast, glucose–insulin–potassium trials yielded inconsistent results,86–88 and increased hospitalisations for HF were documented with thiazolidinediones (e.g. rosiglitazone), incretin-based therapies (e.g. saxagliptin) and sulfonyureas (e.g. glicazide).89–91 Thus, glycaemic control alone might not be sufficient for preventing or managing DiCM.
Because systolic dysfunction develops as DiCM progresses, and can itself accelerate DiCM by independently inducing mitochondrial dysfunction and oxidative stress, the mandated use of HF specific therapies is critical. They not only disrupt the vicious cycle between systolic failure and DiCM but certain agents such as β-blockers directly suppress oxidative stress and FA metabolism.92,93 However, because these agents are not mandated in the majority of DiCM patients (as their LV ejection fractions are adequate), alternative strategies are needed.
Strategies to modulate cardiac substrate utilisation
Increased FA utilisation is a key feature of deranged metabolism in T2DM. Besides statins to reduce cholesterol levels, excessive FA metabolism can also be targeted (Figure 1) by drugs that reduce plasma-free FA levels (lipoprotein lipase inhibitors), mitochondrial FA uptake (CPT1 inhibitors) or FA oxidation (β-oxidation inhibitors). In rats with T2DM, acipimox and etomoxir reduced serum lipid levels and augmented SERCA expression. 94 In patients with T2DM and HF, trimetazidine reduced natiuretic peptides and improved exercise capacity and LV function despite no change in cardiac perfusion. 95 In a large clinical trial, ranolazine relieved angina more in diabetic than in non-diabetic patients. 96 It also recovered LV function quicker in diabetic than in non-diabetic rats after myocardial infarction, with the benefits related to activation of the energy sensor, adenosine monophosphate kinase. 97 Although perhexiline improved cardiac energetics and function in small HF and hypertrophic cardiomyopathy cohorts, it was not beneficial in patients undergoing cardiac surgery and has not been studied in T2DM.98–100 Besides inhibiting FA utilisation, directly stimulating glucose oxidation with dichloroacetate, a PDH activator, could also rebalance cardiac substrate use in T2DM. 101 However, no studies have been done.
Strategies to improve mitochondrial OxPhos
Mitochondrial oxidative stress is implicated in the reduction in ETC activity, and mitochondrial-targeted antioxidants (e.g. SS-31, MitoQ) preferentially accumulate in mitochondria to reduce ROS generation.102,103 In cell models of glucotoxicity and gluco-lipotoxicity, MitoQ reduced oxidative stress, increased OxPhos and promoted survival. 102 Alternatively, oxidative stress could be dampened by boosting antioxidant defences with agents such as resveratrol which improved cardiac OxPhos in a rat model of T2DM. 104 Besides attenuating oxidative stress, direct ETC stimulation could also augment OxPhos.
Electron transfer from reducing equivalents to molecular oxygen is the limiting step in OxPhos. Coenzyme Q10, also known as ubiquinone, carries electrons from their sites of donation (complexes I and II) to complex III and is also a potent antioxidant. In 420 patients with chronic HF, coenzyme Q10 was shown to improve New York Heart Association (NYHA) class and survival. 105 While no studies in patients with T2DM have been done, coenzyme Q10 attenuated LV remodelling and diastolic dysfunction in a mice model of T2DM. 106 Theoretically, iron could also be used to augment OxPhos. It is a functional component of many ETC and Krebs cycle proteins, and its deficiency impairs exercise capacity, induces LV hypertrophy and leads to diastolic and systolic dysfunction.38,39,107,108 Systemic and myocardial iron deficiency is evident in patients with chronic heart failure, is associated with greater exercise intolerance and mortality and its correction improves functional status.107,109–111 To date, iron status in T2DM is largely unknown. Because the rate of PCr regeneration by creatine kinase is also unknown in humans with T2DM, agents that improve energy transduction such as allopurinol and creatine may be relevant once more data are available.
Summary
The metabolic phenotype of the type 2 diabetic heart is characterised by excessive FA utilisation and storage, suppressed glucose oxidation and impaired OxPhos. Myocardial FA excess is posited to play a pivotal role. By inducing steatosis and an abundance of ROS and toxic intermediary metabolites, FAs can trigger adverse cardiac remodelling. By inhibiting key glycolytic enzymes, FAs can shunt glucose towards more pathological pathways that plausibly aggravate remodelling. Further research, particularly in human subjects, is needed to clarify whether defective myocardial metabolism and energetics truly drive unexplained cardiomyopathy in T2DM.
Footnotes
Declaration of conflicting interests
Dr Amaral has no conflicts of interest. Dr Okonko has received research support from Pharmacosmos A/S.
Funding
The authors are supported by the British Heart Foundation. Dr Okonko has also received research support from Pharmacosmos A/S.