
Abstract
Governing a large amount of cellular processes in mammalian cells is a 24-h regulatory mechanism known as the circadian clock. Through the release of neurohormonal factors, the master central clock is able to regulate the otherwise independent peripheral clocks situated in all vital organs. It has recently been shown that forced misalignment of the circadian cycles, often as a consequence of lifestyle factors, is an independent cardiometabolic risk factor and may thus potentially predispose certain groups, such as nightshift workers, to cardiovascular disease. In this review, we will analyse some of the recent advances regarding circadian clock dysfunction and the development of cardiovascular diseases. Finally, we will touch on the developing link between circadian dysfunction and myocardial infarctions.
Introduction
Despite recent decrease in the rates of cardiovascular disease (CVD), this disease was and remains the most common cause of mortality in Europe accounting for 47% of death in the European Union (EU). 1 It is estimated that the cost to the EU economy is around 196 billion Euros. 1 While many risk factors such as smoking, hypertension and hyperlipidaemia have been linked to an increased risk of CVDs, it has only recently been shown that forced misalignment of the behavioural and circadian cycles is an independent cardiometabolic risk factor. 2 Following this discovery, there have recently been some developments in the field of experimental genetics attempting to elucidate this link between the circadian clock present in different tissues and the occurrence of cardiac and metabolic disease.
In this review, we analyse and summarize some of the milestones that have been made in unravelling this still young field of research of circadian rhythm and its influence on the heart.
Circadian rhythm
The diurnal variation of light and dark encompassed in the 24-h day has been known for some time to drive many cyclic changes, not only in humans but in nearly all mammalian cells examined to date. 3 Indeed, the human body is geared to be able to respond to changes in nutrient availability, activity levels and hormonal changes by an internal 24-h clock known as the circadian clock.
This ‘clock’ is composed of two distinctly separated hierarchical pieces. The central clock located in the suprachiasmatic nuclei of the anterior hypothalamus 4 commands a peripheral clock present in most vital organs, including heart, liver, muscle, adipose tissue and kidney (reviewed in Dibner et al. 5 ). While the peripheral clock is mainly influenced through the availability of nutrients (high availability during the day, absent at night) which alter the transcription of key endocrine hormones, 5 the central clock keeps a master control through its release of neurohormonal factors upon sensing of daylight by the retina 6 (Figure 1). The main transcription factors involved in this regulation are brain and muscle aryl hydrocarbon receptor (BMAL1) and circadian locomotor output cycles kaput (CLOCK).7,8 These transcription factors can heterodimerize and activate their downstream targets which include the Period (Per1, Per2 and Per3) and the cryptochrome (Cry1 and Cry2) genes which, further to providing further downstream activation, are also responsible for the system regulation through a negative feedback loop 9 (Table 1). Mice with mutations targeted at either the CLOCK or BMAL1 gene display propensity to develop obesity, hyperglycaemia, hyperlipidaemia as well as increased nitric oxide (NO) synthase uncoupling, suggesting that loss of the normal circadian rhythm may contribute to the development of insulin resistance, obesity and hyperlipidaemia in humans.12,25,26
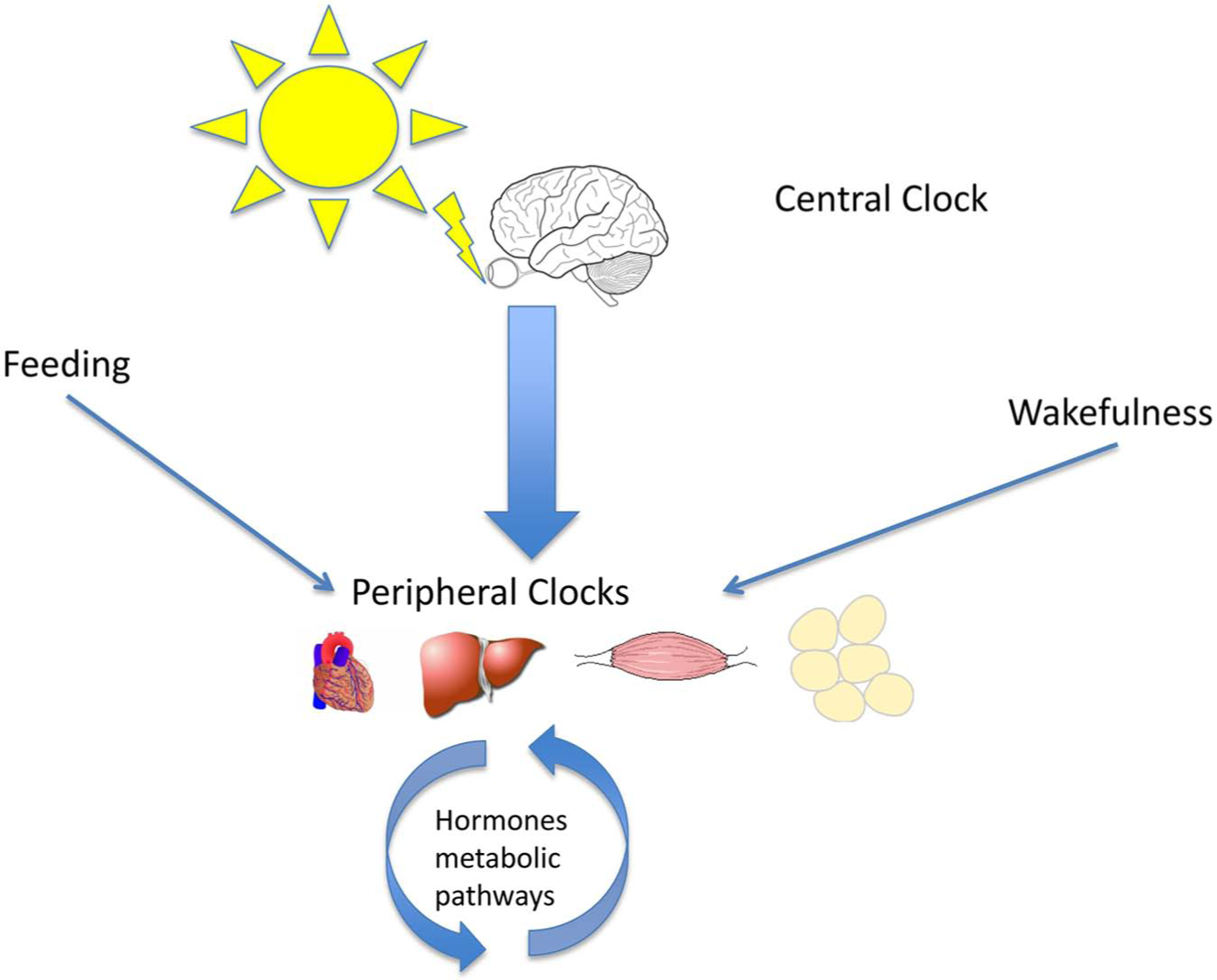
Schematic representation of the circadian clock. The central clock located in the suprachiasmatic nuclei of the anterior hypothalamus 4 is the master switch for the peripheral clocks present in most vital organs including heart, liver, muscle and adipose tissue. The peripheral clock is mainly influenced through the availability of nutrients and the state of wakefulness, which alter the transcription of key endocrine hormones. The central clock keeps a master control through its release of neurohormonal factors upon sensing of daylight by the retina.
Circadian clock genes and their cardiovascular effects.
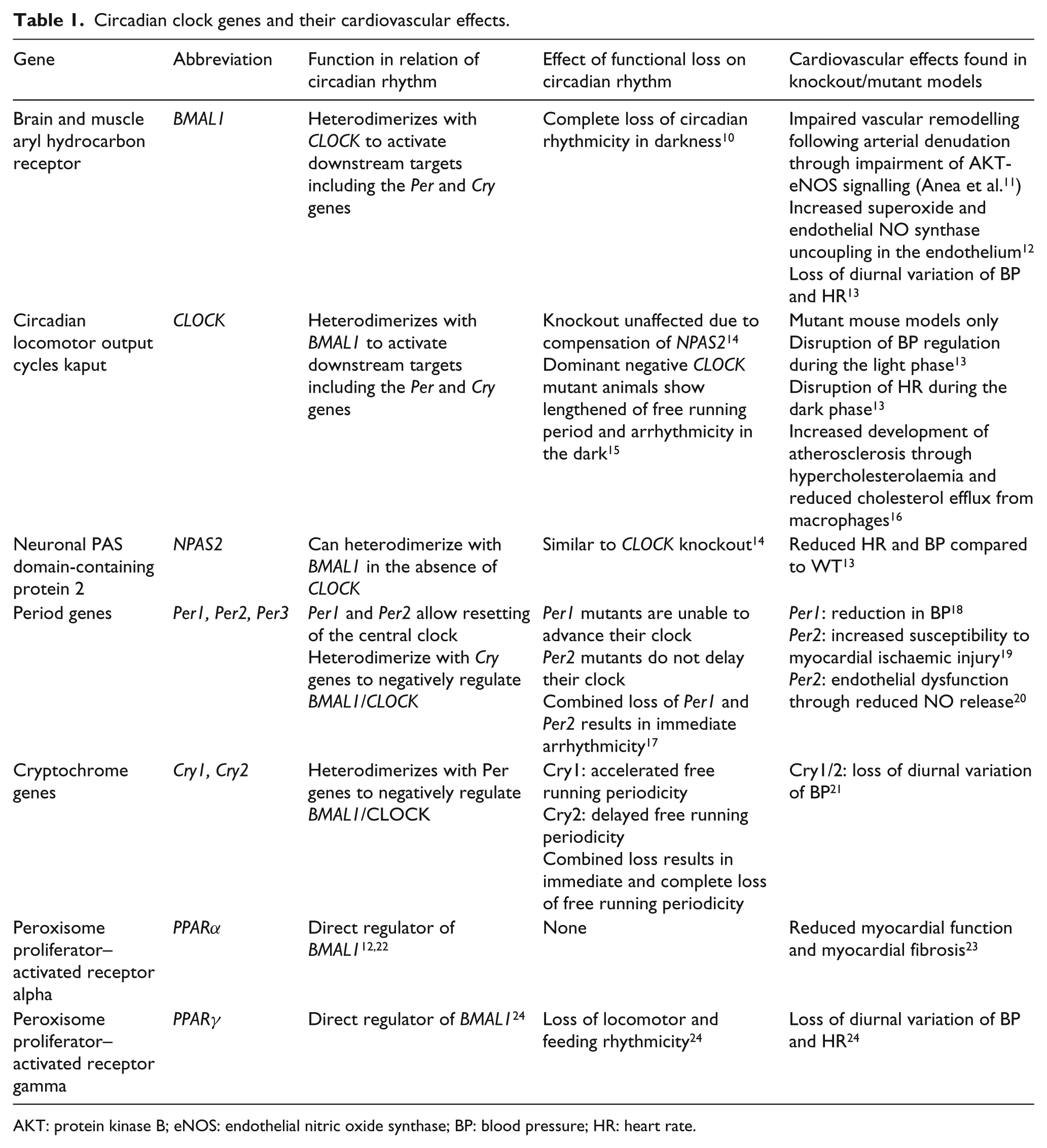
AKT: protein kinase B; eNOS: endothelial nitric oxide synthase; BP: blood pressure; HR: heart rate.
Another important step in unravelling the importance of the internal clock was the demonstration that not only the loss of the central clock elements, such as BMAL1 and CLOCK, but also the loss of their downstream targets, such as PER2, leads to impaired glucose homeostasis and compromised insulin-stimulated NO release independent of obesity in mice. 27
Circadian clock and cardiovascular function
The cardiovascular system is a major system under the control of the circadian rhythm. In order for the heart to function continuously, it requires a constant source of energy. Indeed, the heart does not contain a great reserve of adenosine triphosphate (ATP), and its pool is renewed every 10 s. 28 In the normally functioning heart, ATP is nearly exclusively formed by oxidative phosphorylation (OXPHOS) in the mitochondria. The rate of OXPHOS is balanced to maintain a constant ATP amount by being exquisitely linked to the rate of ATP hydrolysis. 29 This means that despite higher workloads such as during exercise or stress, the heart maintains a constant amount of ATP. 30 To achieve this, the heart requires a constant supply of nutrients. It can be considered as being an ‘omnivore’ as it is able to utilize different energy substrates including fatty acids (FAs), glucose as well as ketone bodies and lactate to generate ATP.
In perfused rat hearts, glucose oxidation rates are increased in hearts isolated during the awake phase with increased expression of GLUT4, increased glycogenolysis and decreased lactate release, 31 thus suggesting that the heart may have a certain anticipatory function regarding energy substrate availability according to daytime (increased feeding during the awake phase and therefore increased availability of glucose during this time).
Lessons from gene knockout models
Much of the current knowledge on the clock system has been gained through the study of flies and rodents and may therefore not be directly applicable to humans since there are some major differences between rodents and humans regarding circadian rhythm (e.g. mice are night active). Furthermore, there are special cases regarding rodents such as hibernation in which case the circadian clock has been shown to stop functioning in some rodents. 32 However, many of the genes involved in the animal clock mechanism (Per1, Per2, Clock, Bmal1) have been described in humans and animal studies and therefore allow some conclusions to be drawn on human mechanisms. 33
BMAL
It was shown that not only cardiomyocytes but also vascular smooth muscle cells (VSMCs) continue to exhibit a diurnal variation in gene expression even when the central clock is removed. 34 Indeed, already some time ago, Keskil et al. 35 showed that aortas taken from rats at different time points showed differing responsiveness to vasoactive agents. McNamara et al. 36 then went on to show that VSMCs continue to exhibit rhythmic oscillation of Bmal1 and Per2 expression when kept in culture. Further investigations into the role of clock genes regarding VSMCs and endothelial dysfunction were carried out by Rudic et al. who investigated the effect of dysfunctional clock genes on the development of vascular diseases. Bmal1-/- and clock mutant mice were analysed, and the authors found that both exhibit pathological response to vascular injury which was, in part, shown to be due to impairment of the protein kinase B (AKT)–endothelial nitric oxide synthase (eNOS) axis leading to endothelial dysfunction (Figure 2). 11 The results were confirmed by the same group using a transplant vasculopathy model which revealed that transplantation of a carotid artery from Bmal1-/- to wild type (WT) leads to an increase in arteriosclerosis, while such an effect is absent when a WT artery is transplanted into a Bmal1-/- mouse 37 (Figure 3).
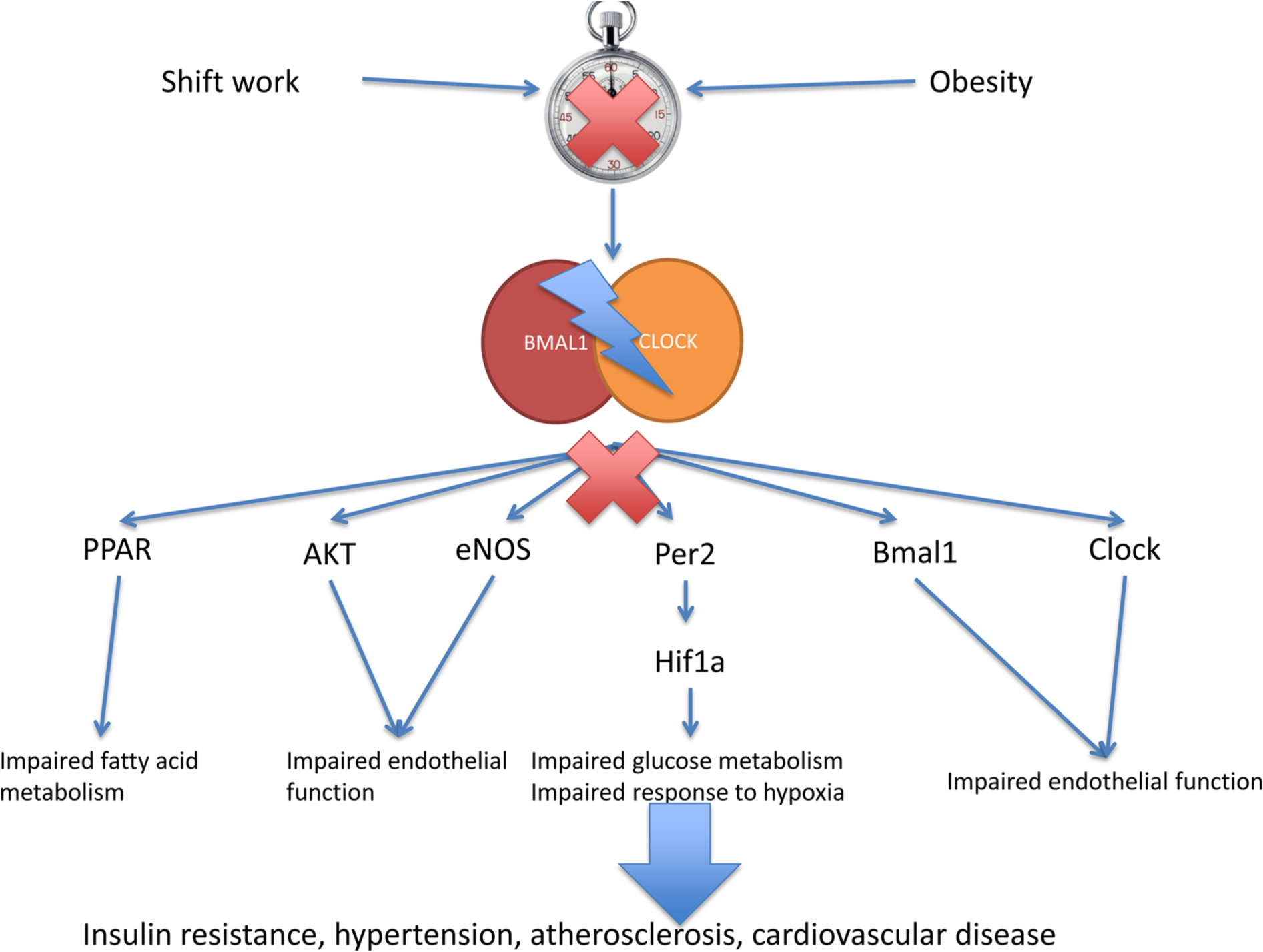
Schematic representation of the effects of circadian dyssynchrony on cardiometabolic function. Impairment of the central or peripheral clock, their dissynchrony or interference with either Bmal1 or Clock gene expression has been shown in vivo in mouse models to interfere with multiple different signalling axes and thus resulting in cardiometabolic dysfunction.
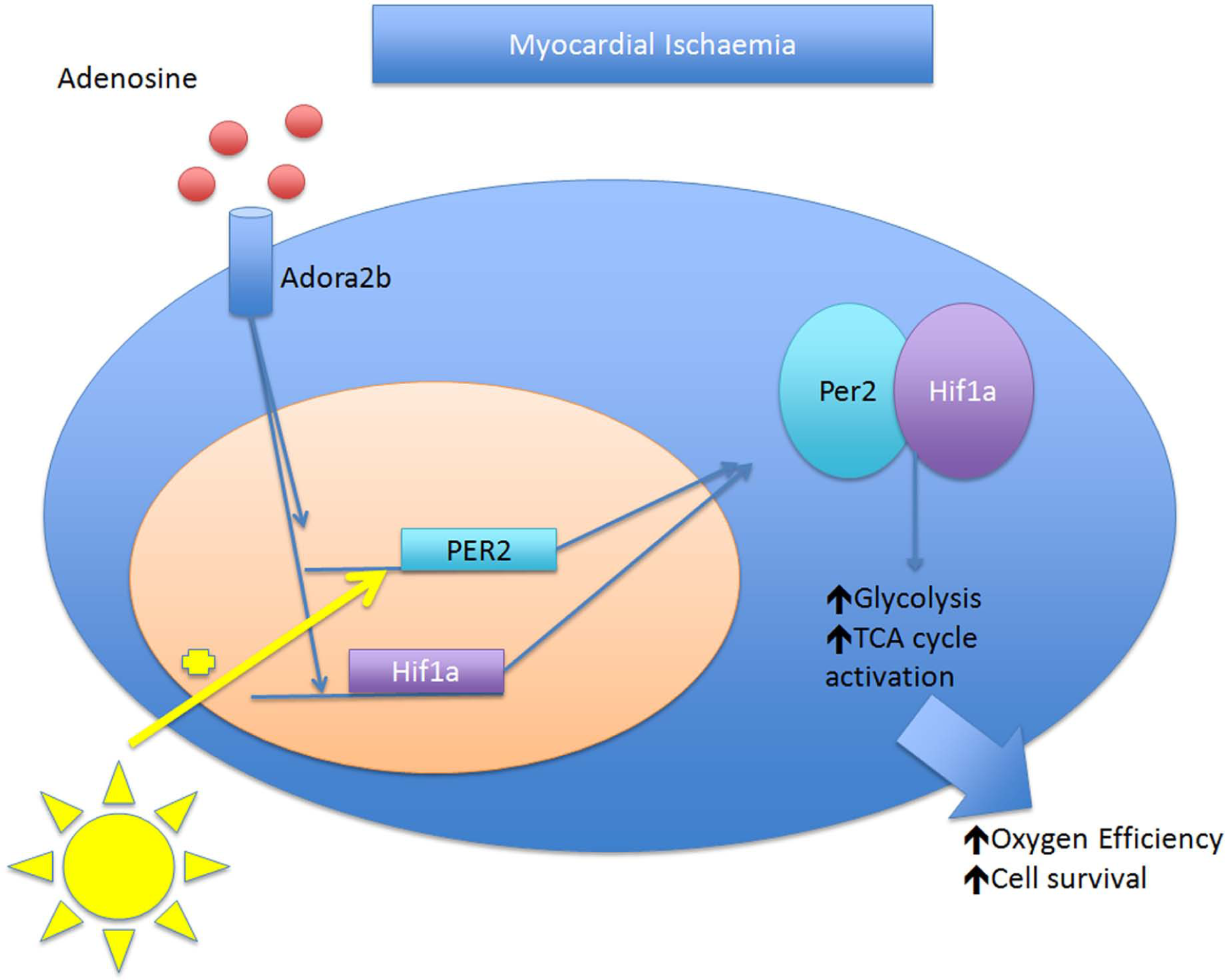
Per2 protects from ischaemic damage through activation of Hif1a. Per2 activation through Adora2b confers protection from ischaemia–reperfusion injury in mice through Hif1a activation resulting in increased glycolysis and thus increased oxygen efficiency.
CLOCK
CLOCK total body knockout mice (Clock-/-) have been found to show no disturbances of circadian rhythm because neuronal PAS domain-containing protein 2 (NPAS2) can substitute for CLOCK deficiency by interacting with Bmal1. 38 However, by deleting the exon 19 of the CLOCK gene (CLOCK19/19), the resultant mutant protein, acting as a dominant negative regulator, disrupts CLOCK function. CLOCK19/19 mice have been shown to exhibit modest hypertryglyceridaemia, hypercholesterolaemia, hyperglycaemia and hyperleptinaemia. 39
The effect of CLOCK function loss on cholesterol metabolism and the development of atherosclerosis have recently been elucidated. Pan et al. 16 found that CLOCK19 mutant protein increases atherosclerosis development in LDLR-/- as well as APOE-/- mice, on a western as well as a normal diet, by increasing plasma cholesterol levels as well as macrophage cholesterol handling.
Mechanistic studies revealed that the CLOCK19 protein enhances cholesterol absorption by enterocytes as well as the uptake of modified lipoproteins by macrophages in the hypercholesterolaemic mouse model ApoE. Furthermore, CLOCK19 macrophages showed reduced reverse cholesterol transport associated with reduced levels of ATP-binding cassette ABCA1. By performing knockdown experiments in WT animals, the other studies showed that CLOCK directly regulates ABCA1 providing evidence that CLOCK dysregulation or dysfunction may directly influence lipid metabolism and macrophage function to enhance the development of atherosclerosis.
Per2
Mice with loss of Per2 function (Per2-/-) were shown to exhibit elevated circulating insulin levels and impaired glucose tolerance tests associated with enhanced glucose-stimulated insulin release and impaired insulin clearance. 40
Initial investigation, gained from PAS domain mutant mice, into the role of Per2 suggested that this may be due to a propensity to develop defective feeding rhythm (increased daytime feeding), impaired glucocorticoid rhythm as well as overall hyperphagia. 41 However, the model used in these studies did not represent a real loss of function model, and recent evidence has refuted earlier result by showing that whole-body knockout Per2 animals exhibit normal feeding and behavioural patterns. 42
Further to the effects of Per2 on glucose metabolism, there has recently been compelling evidence suggesting that Per2 is able to directly influence lipid metabolism by interacting with PPARγ and repressing its transcriptional activity in white adipose tissue. Indeed, lipidomic analysis revealed an increase in the oxidation rate of FA in Per2-/- mice, with a concomitant reduction in fat pad size and adipocyte diameter. 42 Interestingly, the repressive activity of Per2 on PPARγ is different from its repression on the CLOCK:BMAL1 complex in that it is not only CRY independent but also tissue specific, being restricted to the WAT without any effects in the liver or brown adipose tissue. This underlines the likely existence of independent pathways of PER2 function, with a circadian clock–independent mechanism of repression of PPARγ as well as the circadian-dependent CLOCK:BMAL1 inhibition. 42
The intimate involvement of Per2 and its role in cardiac metabolism during myocardial ischaemia were recently elucidated by Eckle et al. 19 who showed that Per2 activation through adenosine receptor A2b (Adora2b) confers protection from ischaemia–reperfusion (I/R) injury in mice. The authors showed that during I/R, Per2-/- mice showed impaired glycolysis and thus impairment of the oxygen efficient pathway in the face of ischaemia, with increased uncoupling of glycolysis during the reperfusion suggesting mitochondrial dysfunction. The authors linked this effect to a Per2-dependent hypoxia-inducible factor 1-alpha (Hif1a) activation. Interestingly, Hifa1 displays a similar diurnal variation in the cardiac tissue as Per2. 19
Hif1a is a key transcriptional element in the glycolytic pathway as well as a strong response element protecting from hypoxia. 43 Its activation has been shown to protect from ischaemia by increasing neovascularization through the upregulation of vascular endothelial growth factor and angiopoietin 1 and 2, 44 the recruitment of endothelial progenitor cells to the affected areas, 45 the preconditioning of tissue to ischaemic insults 46 and the upregulation of glycolysis as an oxygen-sparing energy pathway. 43
It was shown that infarction size in rodents varies according to daytime and that a high Hif1a expression in the hours 12–18 (late evening) resulted in smaller infarction sizes. Thus, as discussed above, Per2 expression may contribute to myocardial protection from ischaemia by upregulation of the oxygen efficient pathway of glucose utilization through the upregulation of Hif1a.
PPARγ
Further to its repression by Per2 in the WAT, there appears a role for PPARγ as a body-wide key integrator of molecular clocks. It was shown that PPARγ exhibits a strong diurnal variation, and in mice, altering the light/darkness cycle of the time of feeding can alter its expression. Mice lacking PPARγ in the vasculature develop disturbances in the diurnal variation of blood pressure (BP) and heart rate (HR) as well as a reduction in the variation in urinary norepinephrine/epinephrine excretions similar to that seen in Bmal1-/- mice. 24 Indeed, animals fed Rosiglitazone, a PPARγ agonist, showed induction of Bmal1 in the aorta. There is some evidence that this is also applicable to humans as pioglitazone, another PPARγ agonist, can restore normal circadian variation of BP. 47
This phenotype was confirmed in animals with whole-body knockout of PPARγ. These displayed a striking loss of rhythmicity, regarding not only HR and BP but also locomotor activity and feeding behaviour. Further to this, PPARγ null mice also showed loss of peripheral circadian rhythmicity of gene expression patterns of clock genes (BMAL1, Per1, Per2, Per3, Cry1) in the liver and WAT. 48
PPARα
PPARα is part of the superfamily of nuclear receptors responsible for the regulation of lipid metabolism. Being sensitive to the availability of FAs, it is responsible for the heart’s rapid ability to adapt to circulating energy substrates. 49
For some time now, it has been known that the expression of PPARα follows a diurnal rhythm in mice and rats. 50 While this may, in part, be explained by the hormonal influence of glucocorticoids and insulin that directly affect transcriptional activity of PPARα, it has been shown that it is regulated in a clock-dependent manner in several peripheral tissues. 51
The first evidence regarding the interaction between PPARα and the clock mechanism came from the observation that obese mice, which lost weight, showed upregulation of PPARα with a concomitant restoration of diurnal variation of HR and BP. 52 Similarly, when diabetic patients were treated with fenofibrate, a PPARα agonist, there was a marked antihypertensive effect at night that was not reflected in overall daytime BP. Furthermore, fenofibrate lowered the average HR over the whole 24-h day, 53 thus suggesting a potential direct link between PPARα and the clock mechanism.
While PPARα knockout mice (PPARα-/-) were shown to exhibit normal circadian locomotor and unaffected SCN clock gene expression, they do exhibit a loss of rhythmicity in the liver transcription profiles of Bmal1 and Per3. 22 Conversely, fenofibrate, a PPARα agonist, is able to upregulate the expression of Bmal1 in the liver suggesting a direct link between PPARα levels and Bmal1 expression. Indeed, there appears to be a direct regulation of Bmal1 transcription by PPARαs through its ability to bind to the promoter of Bmal1. At the same time, Bmal1-/- mice show a dysregulation of PPARα rhythmicity demonstrating a feedback loop wherein PPARα promotes Bmal1 transcription which in turn supplies a circadian regulation on PPARα. 22
To summarize, we can say that while research into the exact mechanism in which clock genes interact with cardiovascular metabolism is still in its infancy, there is no denying that there is a definite link which will need to be explored further.
Circadian clock and myocardial infarction
The observation that myocardial infarctions are increased in the early hours of the morning was already made over 20 years ago. 54 A meta-analysis by Cohen et al. 55 including over 60,000 acute myocardial infarction (AMI) patients showed a strongly increased incidence in the hours between 06:00 a.m. and midday. This increased mortality was previously mainly thought to be associated with increased sympathetic activity in the waking phase. Indeed, administration of a beta-blocker resulted in the loss of the morning excess of AMI. 56 However, recent discoveries have shed a new light on this topic.
A perfect population to study the mechanism of clock disruption and its effects includes shift workers in whom the risk of developing obesity,57,58 diabetes 59 or premature death is greatly increased. 60 The circadian misalignment seen in this group has been shown to correlate with insulin resistance, cortisol derangements as well as increased BP. 2
When examining the Swedish Registry for AMI which keeps accurate data of all AMI in Sweden since 1987, Janszky and Ljung 61 found that the incidence of AMI is also increased in the first 3 days after the change from winter to summer time. On the contrary, rates of AMI were reduced when switching from summer to wintertime, which adds an hour of sleep. This trend was confirmed by Jiddou et al. 62 in a retrospective study of 935 patients carried out between 2006 and 2012 in Michigan.
While the authors present some valid reasoning for this increased occurrence, including acute sleep deprivation–induced sympathetic predominance and catecholamine release, no single mechanism has been elucidated to date accounting for this phenomenon. 63
The incidences of myocardial infarction as well as the onset of arrhythmias have also been shown to be increased during the switch from the sleep to the awake state. Several mechanisms in the heart confer protection from ischaemia, and the survival of ischaemic cardiomyocytes, in part, depends on the activation of these pathways. 64 Intriguingly, rather than being constantly expressed, some of the proteins involved in these pathways appear to show a circadian dependence. 65 Durgan et al. 66 recently showed that mice undergoing I/R injury at the sleep-to-wake transition exhibited greater infarction sizes than mice that underwent I/R at the wake-to-sleep transition moment. These changes were abolished in animals with genetic ablation of the circadian clock in the cardiomyocytes. The authors linked this to the circadian variations in the phosphorylation of the pro-survival genes AKT and GSK-3β.
So far, very few groups have attempted to reproduce the animal data in humans, and the data obtained remain somewhat controversial. Suarez-Barrientos et al. 65 retrospectively analysed 811 ST-elevation myocardial infarction (STEMI) patients admitted in a single centre in Spain. The authors found a clear association between time of day and incidence of ischaemic events and also infarction size as determined by peak creatine kinase and troponin I. In this patient group, infarction size was found to be larger in the patient group with onset of symptoms between 06:00 a.m. and noon.
Reiter et al. 67 similarly retrospectively analysed 165 STEMI patients and found that myocardial injury was greatest at 01:00 a.m. onset of ischaemia and 05:00 a.m. onset of reperfusion, even when adjusted for area at risk as measured by cardiac magnetic resonance imaging (MRI) and angiographic assessment.
While there is a slight discrepancy between the data obtained regarding peak time of myocardial ischaemic damage, in part, probably due to Reiter et al. retrospectively excluding a large number of patients (around 85%) rather than including and analysing all patients in an unbiased way and then adjusting for potential bias, the clear trend regarding daytime-dependent myocardial injury size appears obvious.
However, earlier this year, Ammirati et al. 68 through the retrospective analysis of data from the First Acute Myocardial Infarction (FAMI) study, carried out between 2002 and 2007 in three different countries (Italy, China and Scotland), while confirming the increased incidence of myocardial infarction in the early hours of the morning, were unable to reproduce the effect of time of day on infarction size seen by Suarez-Barrientos et al. and Reiter et al. despite multiple statistical models.
The ambiguousity of these results shows that more research into the exact molecular mechanisms underlying infarction size and daytime will need to be carried out.
Perspectives
Taken together, these data suggest that disturbance of the circadian clock may not only contribute to metabolic changes such as insulin resistance and diabetes but also may directly influence CV risk and the occurrence of cardiovascular events. However, a better understanding of the underlying molecular mechanisms is needed for the development of diagnostic and eventually therapeutic strategies. What we are lacking are, however, reliable markers to estimate how the lifestyle is impacting on circadian regulation. Investigating subjects with a misaligned circadian rhythm such as shift workers may lead to the identification of novel biomarkers of the disturbed clock system and thus help us to gain more insight into the role of the circadian clock in the cardiovascular system.
Footnotes
Declaration of conflicting interests
The authors declare no conflicts of interest.
Funding
This work was funded, in part, by FP-7 EURHYTHDIA, Fondazione Roma 2008, ESFD/Lilly 2012, AIRC 2012 Project IG 13163, FP7-Health-241913 FLORINASH, PRIN 2009FATXW3_002 and SID Grant 2013 to Massimo Federici. Robert Stöhr was supported by a fellowship from the Deutsche Herzstiftung and the University of Rome Tor Vergata.