
Abstract
Resistin, an adipokine associated with the metabolic syndrome, is believed to have a role in thrombotic conditions. This work analyses the effects of resistin on P-selectin expression using a combination of ex vivo human studies, in vivo animal models and in vitro cell cultures. Human platelets and vascular endothelial cells were incubated with resistin, with or without anti-Toll-like receptor 4 (TLR-4) or mitogen-activated protein kinases (MAPK) pathway inhibitors, whereas mice were treated with resistin infusion followed by analysis of P-selectin expression. Resistin increased both human and murine platelet P-selectin expression compared with controls (human: 48.02% ± 7.6% vs 35.12% ± 2.62%, p < 0.05; mouse: 8.17% ± 0.37% vs 4.44% ± 0.37%, p < 0.05), through the p38 MAPK pathway. In contrast, resistin had no effect on endothelial P-selectin production. We conclude that resistin induces platelet activation by increasing P-selectin expression through the p38 MAPK-dependent pathway. These data provide one mechanism for the prothrombotic state in individuals with the metabolic syndrome.
Introduction
Metabolic syndrome is a cluster of cardiovascular diseases risk factors, including abdominal obesity, hypertension, insulin resistance and dyslipidemia. 1 Several studies have shown close associations between the metabolic syndrome and venous or arterial thrombosis. 2 Plasma levels of resistin, an adipocytokine believed to antagonize insulin action, are elevated in individuals with the metabolic syndrome. 3 Although resistin expression was initially defined in adipocytes, significant levels of protein expression are also found in monocytes, macrophages, spleen and bone marrow cells. 4 More importantly, high plasma levels of resistin are associated with vascular thrombotic conditions, including acute coronary syndrome. 5 This suggests that resistin is not only associated with the metabolic syndrome, but also plays an important role in the development of vascular thrombotic complications.
P-selectin is synthesized in both platelets and endothelial cells, and it is little expressed in quiescent platelet, but could be quickly released when platelets are activated by physiological or pathological agonists and helps to maintain platelet activation. 6 To date, the direct effect of resistin on P-selectin expression has not been reported.
In this study, we examined the effects of resistin on platelet P-selectin expression using ex vivo human and in vivo animal studies, supplemented by in vitro cell culture work. Moreover, we studied the signalling pathways that may be involved in resistin-mediated modulation of platelet activation.
Methods
Study participants
Healthy human volunteers (n = 8) were recruited from Medical Examination Center of the First Affiliated Hospital of Shantou University Medical College. Washed platelets were collected and were treated with different concentrations of resistin (25, 50, 100 and 200 ng/mL), with or without thrombin (0.05 U/mL), or preincubated with anti-Toll-like receptor 4 (TLR-4) antibody (1 µg/mL), PD98059 (10 µM) or SB203580 (10 µM; Sigma, St Louis, MO, USA) for 15 min before resistin treatment.
Mouse model
Male C57BL/6J mice (Vital River Laboratory Animal Company, Beijing, China) were housed in a temperature-controlled animal facility in Shantou University Medical College. Mice were randomized into saline group (n = 5) and resistin group (n = 5) by infusion of saline or resistin (Perprotech, Rocky Hill, NJ, USA) (33 µg/kg/day) for 14 days. Washed platelets were collected for P-selectin assays in basal state and after thrombin activation (0.05 U/mL).
Detection of platelet P-selectin expression
P-selectin levels were measured by flow cytometry. We applied CD61-fluorescein isothiocyanate (FITC) (human) or CD41-FITC (mouse), CD62P-PE or isotype-matched control IgG1-PE (eBioscience, San Diego, CA, USA) for P-selectin assay on a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA).
Cell culture and detection of endothelial P-selectin expression
Primary human umbilical vein endothelial cells (HUVEC) (ScienCell Research Laboratories, San Diego, CA, USA) were incubated with various concentrations of resistin (25, 50 and 100 ng/mL) for different times (6, 24 and 48 h). Endothelial cell–derived P-selectin levels were measured by flow cytometry.
Statistics
Data are expressed as mean ± standard deviation (SD), and statistical evaluation was performed using one-way analysis of variance (ANOVA) and the unpaired two-tailed t-test. A value of p < 0.05 was considered statistically significant.
Results
General characteristics of participants
Participants include four males and four females; their characteristics are described in the supplementary data.
Resistin increased human platelet membrane P-selectin levels
Resistin treatment resulted in increased P-selectin expression in a dose-dependent manner, with the maximum effect being reached with 50 ng/mL (48.02% ± 7.6% vs 35.12% ± 2.62%, p < 0.05). P-selectin expression was higher upon co-incubation with thrombin and resistin compared with thrombin treatment alone (50.41% ± 3.59% vs 45.08% ± 2.7%, p < 0.05) (Figures 1 and 2).
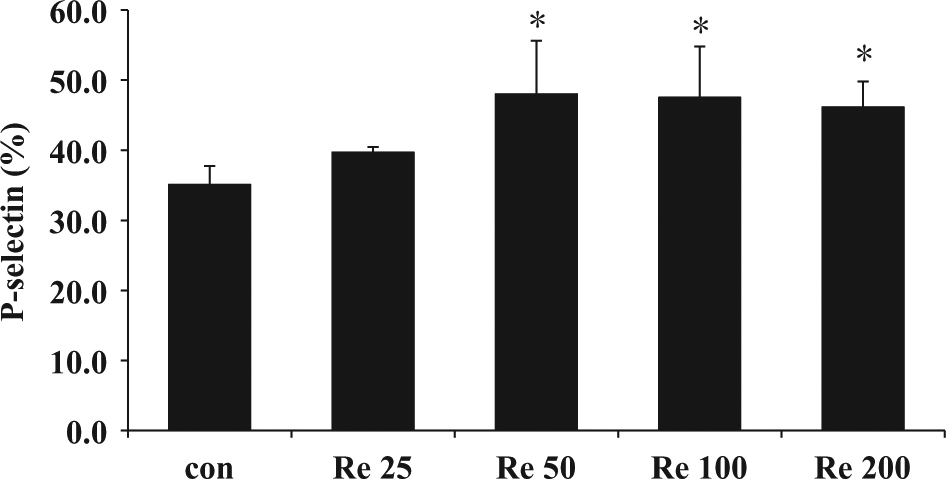
Effect of resistin on human platelet P-selectin expression. Platelets from human blood were stimulated with resistin (25, 50, 100 and 200 ng/mL). *p < 0.05 versus control.
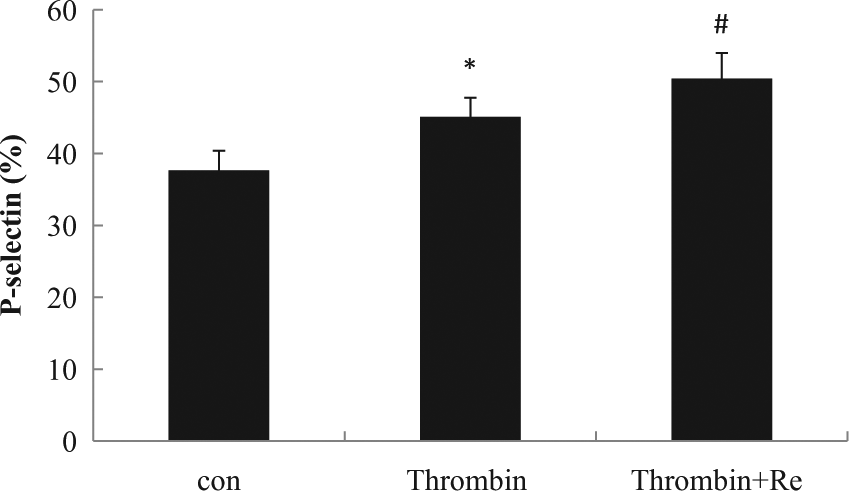
Effect of resistin on thrombin-induced platelet P-selectin expression. Washed platelet from human blood was stimulated with thrombin (0.05 U/mL), with or without resistin (50 ng/mL) for 15 min. *p < 0.05 versus control; #p < 0.05 versus thrombin-treated group.
Pretreatment with the p38 mitogen-activated protein kinases (MAPK) inhibitor SB203580, but not extracellular regulated protein kinases (ERK) inhibitor PD98059 or anti-TLR-4 antibody, attenuated the effects of resistin on platelet P-selectin expression (Figure 3).
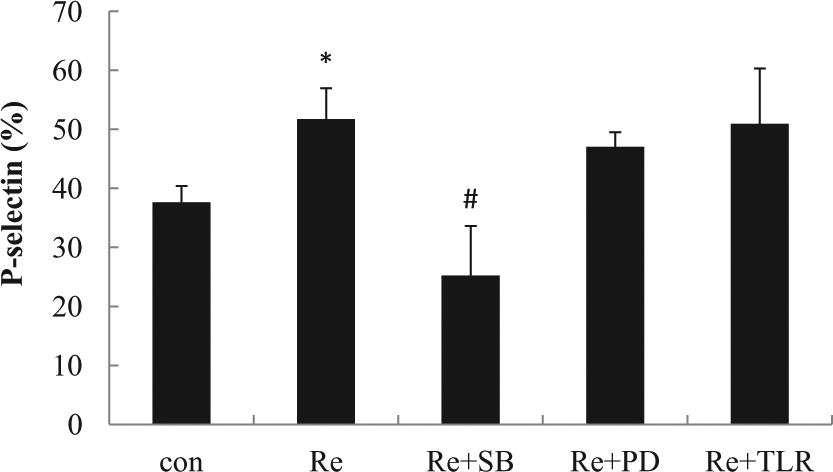
Role of TLR-4, p38 MAPK and ERK pathways on resistin-induced platelet P-selectin expression. Washed platelet from human blood was preincubated with anti-TLR-4 antibody (TLR, 1 µg/mL), p38 MAPK inhibitor SB203580 (SB, 10 µM) or ERK inhibitor PD98059 (PD, 10 µM) for 15 min; then treated with resistin (50 ng/mL) for 15 min. *p < 0.05 versus control; #p < 0.05 versus resistin-treated group.
Resistin increased platelet P-selectin in mice
Resistin enhanced basic murine platelets P-selectin expression significantly compared to those treated with saline (8.17% ± 0.37% vs 4.44% ± 0.37%, p < 0.05) (Figure 4). P-selectin levels were higher upon co-incubation with thrombin and resistin compared with thrombin treatment alone (15.25% ± 0.75% vs 10.02% ± 0.9%, p < 0.05).
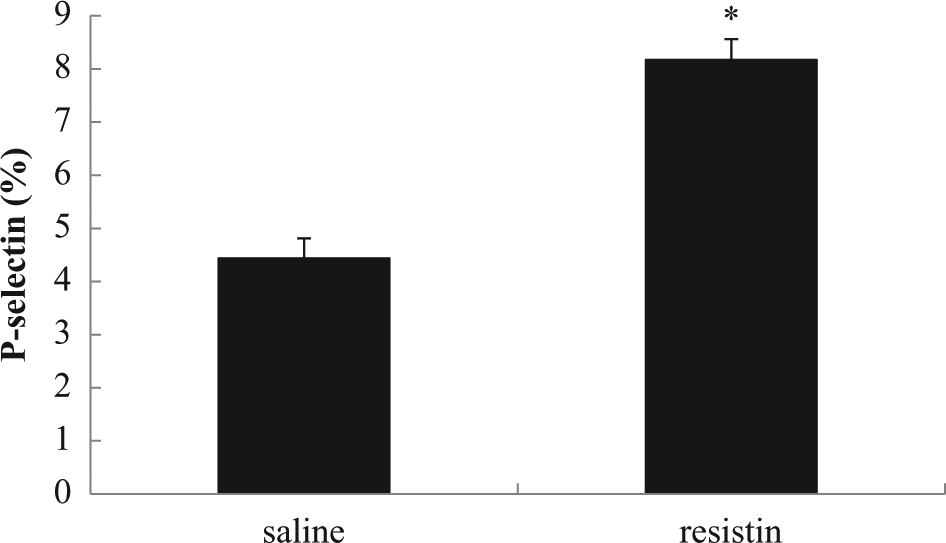
Effect of resistin on platelet P-selectin expression in mice. C57BL/6 mice 12 weeks old were injected with resistin (33 µg/kg/day) or saline through intraperitoneal injection for 2 weeks. *p < 0.05 versus saline group.
Resistin did not influence endothelial P-selectin
P-selectin expression was not detected on HUVEC cells using different resistin concentrations (25, 50 and 100 ng/mL) at various time points (6, 24 and 48 h; Figure 5).
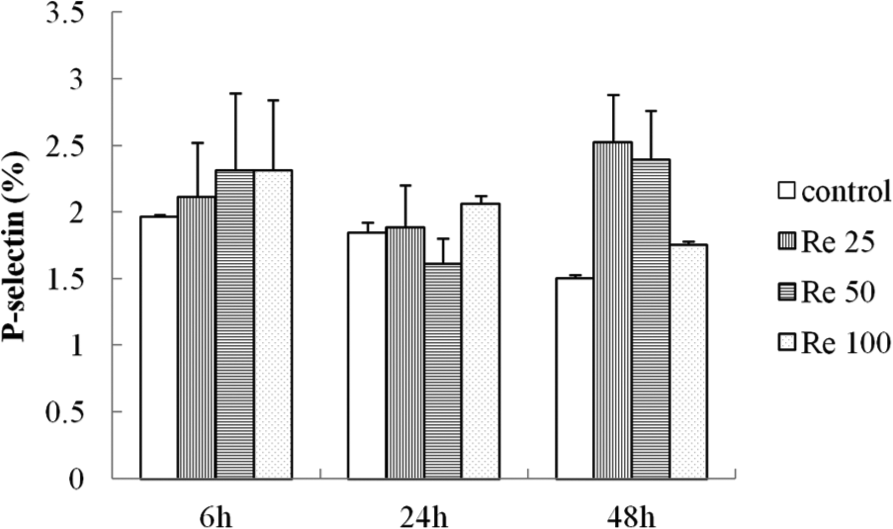
Effect of resistin on endothelial P-selectin expression in HUVEC. HUVEC were incubated with media only or different concentrations of resistin (25, 50 and 100 ng/mL) for different times (6, 24 and 48 h).
Discussion
The results of our study demonstrate that resistin directly stimulates human and mouse platelet P-selectin expression via p38 MAPK pathway, an effect that is enhanced by thrombin. This suggests that resistin plays a role in platelet activation and thrombus formation.
Individuals with metabolic syndrome are reported to have a higher risk of thrombotic events. 7 Platelets activation, which initiates thrombus formation, is considered to be one of the major risks for prothrombotic state in metabolic syndrome patients. 8 As a marker of platelet activation, P-selectin is associated with cardiovascular disease by inducing platelet–platelet and platelet–leukocyte aggregations. 6 Resistin showed an effect on platelet stimulation independently of thrombin providing a new mechanism for platelet activation in individuals with high levels of this adipokine. We also found that resistin enhanced thrombin-induced P-selectin expression, suggesting a synergistic or at the very least an additive effect for resistin in thrombin-induced platelet activation. In our in vitro study, resistin did not increase vascular endothelial P-selectin expression, although a trend was observed after resistin incubation for 48 h. Other studies, however, demonstrated that resistin increased P-selectin expression in endothelial cells. The discrepancy may be related to different characteristics of the HUVEC used or may be due to subtle methodological differences. 9
We have shown that inhibition of p38 MAPK, but not ERK, attenuated resistin-induced P-selectin levels elevation, indicating that procoagulant intracellular signals elicited by resistin are likely to be mediated through the p38 MAPK signalling mechanism. This observation is consistent with other studies showing a similar role for the p38 signal pathway in P-selectin regulation in platelet. 10
In summary, our study demonstrates that resistin contributes to platelet activation by directly increasing platelet P-selectin levels through the p38 MAPK signal pathway. This further helps to understand the mechanisms for the prothrombotic state in individuals with the metabolic syndrome or those with diabetes. Furthermore, our data may provide an explanation for the reduced efficacy of antiplatelet agents in individuals with diabetes.
Our study has a number of limitations: it is unclear whether increased platelet activation by resistin is directly responsible for increased platelet aggregation, and this requires further investigation. Also, it remains to be shown whether platelet activation by resistin has an adverse effect on vascular thrombus formation in vivo. Work is currently ongoing to clarify the above points, which will further help to understand the role of resistin as a potential anti-thrombotic therapeutic target.
Footnotes
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Funding
This project was funded by grants from the Guangdong Provincial Science & Technology Foundation (grant number 2009B030801325 to HJ), the Medical Scientific Research Foundation of Guangdong Province (grant number B2011218 to WQ) and the National Natural Science Foundation of China (grant number 81070413 to HJ).