
Abstract
Haemophagocytic lymphohistiocytosis (HLH) is a rare, aggressive, excess immune activation syndrome. Diagnosis can be challenging due to its several clinical mimics including sepsis. There are multiple aetiologies of HLH; in adults, it is most commonly triggered by infection, malignancy, drugs and autoimmune processes. Failure to rapidly diagnose and treat this condition can be fatal. The management of HLH includes identifying and removing the trigger, supportive management and immunosuppression. Identifying the trigger is essential to inform the most appropriate type of immunosuppression. Here, we report a case of likely drug-induced HLH in a patient recently treated for hairy cell leukaemia. The culprit drug was thought to be co-trimoxazole and this case report highlights a very rare complication of this commonly used drug. We discuss our management approach with steroid monotherapy and withdrawal of co-trimoxazole.
Keywords
Introduction
Haemophagocytic lymphohistiocytosis (HLH) is an aggressive and life-threatening syndrome characterised by excess immune activation. 1 HLH can be primary (genetic) or secondary (acquired). Secondary HLH accounts for most adult cases and can be triggered by malignancy (lymphomas), rheumatological disorders (macrophage activation syndrome), infections (typically viral), metabolic disorders or drugs. 2 If left untreated, HLH carries a mortality of over 90%. However, the presentation of HLH can be mimicked by other conditions, particularly sepsis, and therefore it can be easily misdiagnosed.
Case
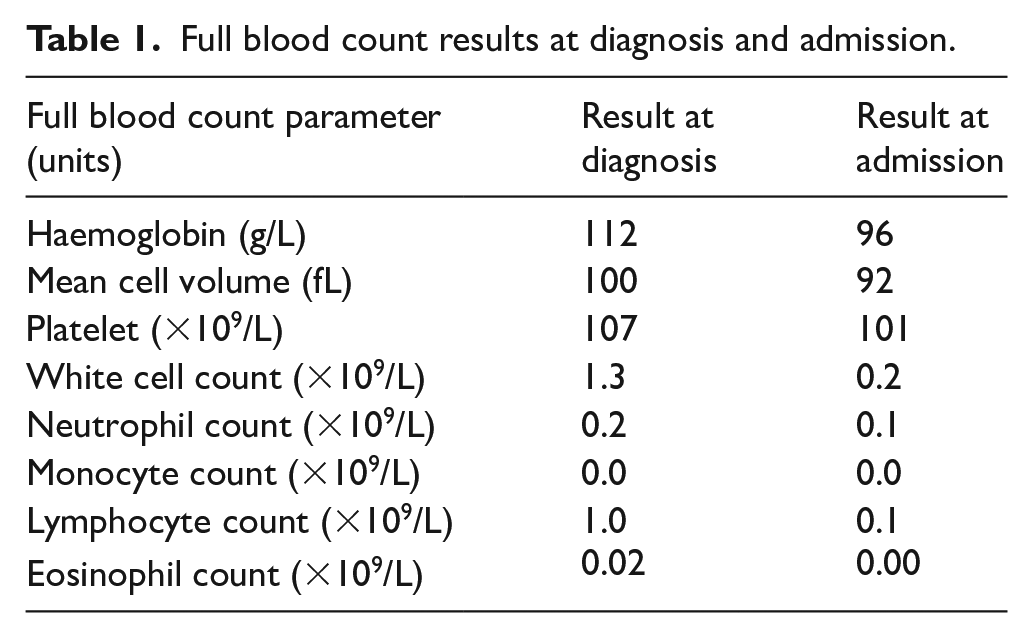
A 45-year-old male with no significant past medical history presented to his general practitioner with a 1 month long history of night sweats and unintentional weight loss. A full blood count revealed pancytopenia (Table 1). A diagnosis of hairy cell leukaemia was made based on peripheral blood film and bone marrow aspirate morphology, immunophenotype, genetics and bone marrow trephine biopsy. Baseline imaging with computed tomography (CT) revealed a 16 cm splenomegaly. As per local and national guidelines, 3 he was treated with five consecutive days of subcutaneous cladribine (0.14 mg/kg) and prophylactic anti-infective agents (acyclovir and co-trimoxazole). Prophylactic granulocyte colony-stimulating factor (G-CSF) was not initiated at this time as per local policy.
Full blood count results at diagnosis and admission.
Ten days after completion of cladribine, the patient developed pyrexia and mild headache. He was admitted to the hospital for the management of presumed neutropenic sepsis with broad-spectrum intravenous (IV) antibiotics. However, there were no localising features of infection and microbiological investigations including peripheral blood cultures, viral throat swabs and urine cultures did not isolate any pathogenic microorganisms. The admission chest X-ray was normal. Due to ongoing high-grade pyrexias (>40°C), antibiotic coverage was broadened, and antifungal cover was added with IV caspofungin. CT imaging revealed only mild peribronchial wall thickening in the lung bases and stable splenomegaly.
In addition, G-CSF was commenced and further assessment for infectious agents was performed, all of which proved negative; an echocardiogram revealed no valvular abnormalities and an induced sputum was non-productive. Due to poor venous access, a central venous line was inserted and the antimicrobial coverage was broadened with IV teicoplanin to cover gram-positive organisms. G-CSF was discontinued due to the development of bony pains.
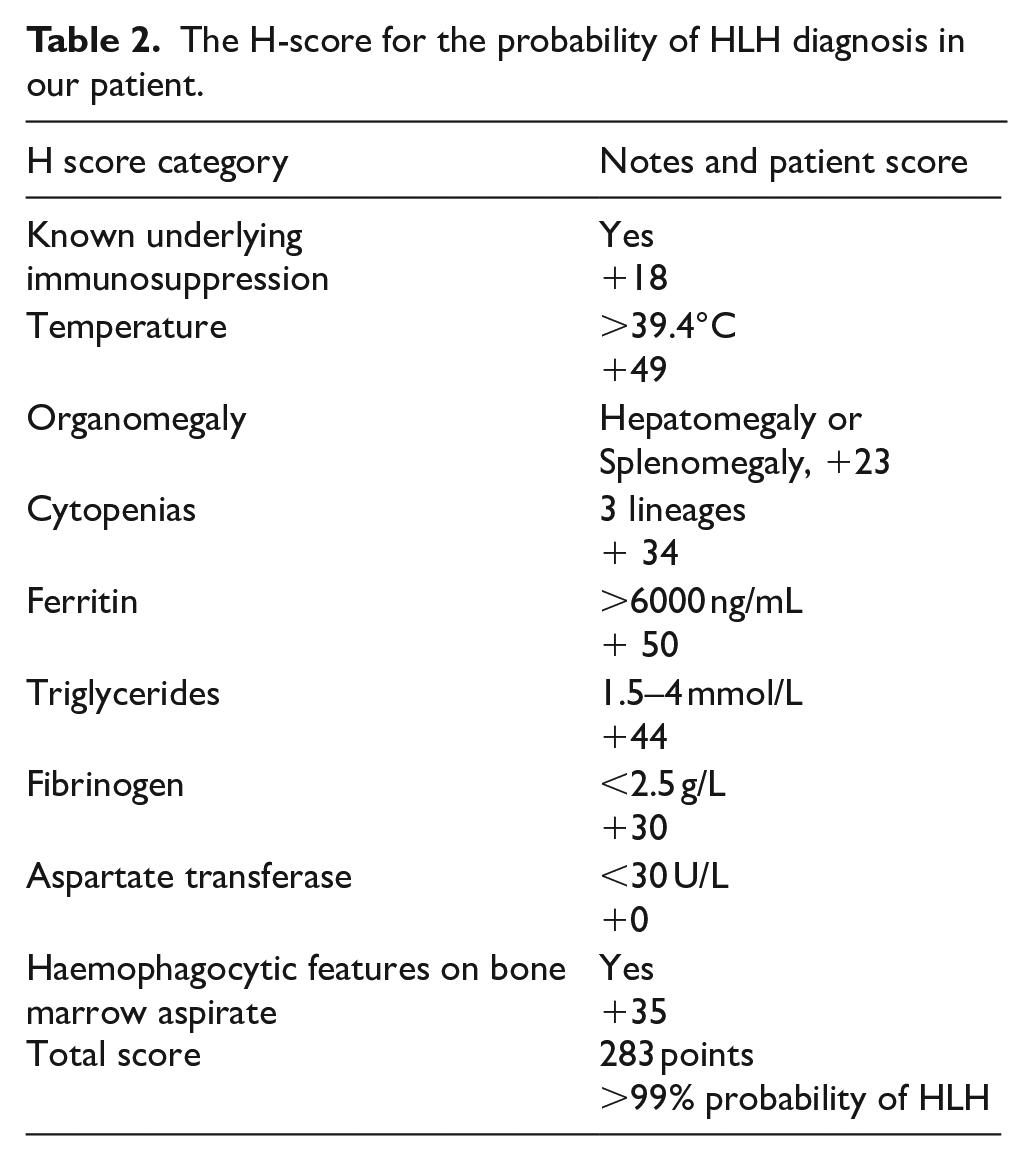
On day 5 following admission, there was a further deterioration in both clinical and laboratory parameters, including the development of pulmonary infiltrates on chest X-ray and a new oxygen requirement (Figure 1). High-dose IV co-trimoxazole was added to cover for pneumocystis jirovecii pneumonia (PJP) and disseminated intravascular coagulation (DIC) was managed with cryoprecipitate and fresh frozen plasma. A ferritin level was reported at the institutional upper limit of detection – >33,511 µg/L. A diagnosis of HLH was considered probable (99% probability) based on the patient’s H-score (Table 2). 4 A repeat bone marrow trephine showed changes consistent with this diagnosis (Figure 2).
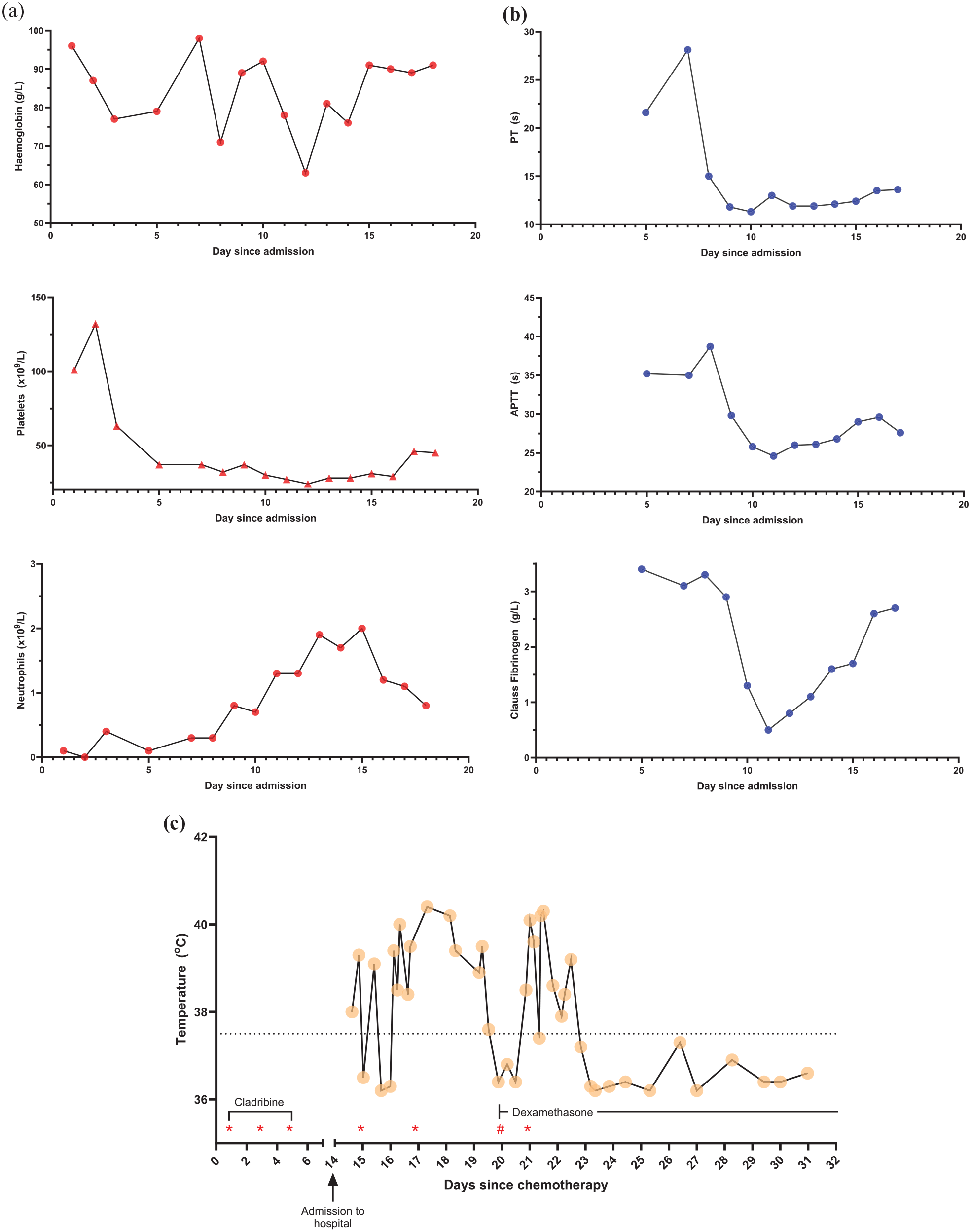
(a) Progressive changes in full blood count parameters through admission. Local reference ranges – haemoglobin 140–180 g/L; platelet 140–400 × 109/L; neutrophils 1.5–7.0 × 109/L. (b) Progressive changes in coagulation parameters through admission. Local reference ranges – prothrombin time 10.1–13.1 s; activated partial thromboplastin time 21.0–30.6 s; Clauss fibrinogen 1.7–4.2 g/L. (c) Progressive changes in temperature (°C) measurements throughout hospital admission and associated timeline of relevant medications.
The H-score for the probability of HLH diagnosis in our patient.
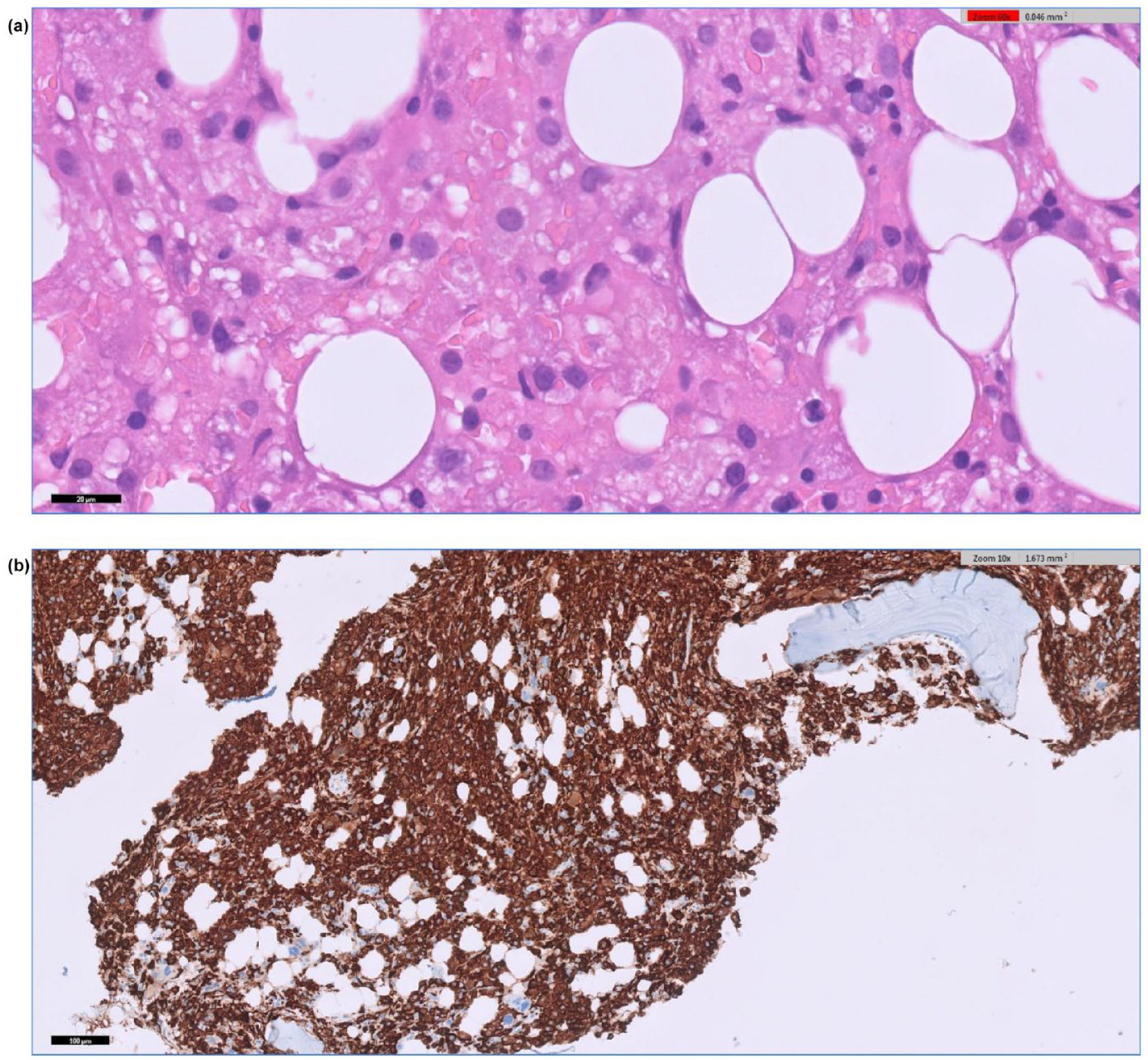
(a) Bone marrow trephine biopsy (60× magnification): the haematopoietic elements are replaced by a diffuse infiltrate of phagocytic cells showing red blood cell Emperipolesis. (b) CD163 immunohistochemistry (10× magnification) highlights the abnormal diffuse phagocytic infiltrate in the interstitial and peritrabecular space.
An assessment for secondary HLH triggers was performed. This included viral screening for cytomegalovirus, Epstein-Barr virus and adenovirus which were negative. No lumbar puncture was performed due to the patient’s DIC, thrombocytopenia and lack of clinical evidence of meningitis.
Antimicrobials were stopped and the patient was commenced on 10 mg/m2 dexamethasone. Anakinra, an interleukin-1 receptor antagonist, was considered, 5 but not administered given the patient’s dramatic clinical improvement. Although there was a slight worsening in his liver transaminases, these steadily improved. The patient was discharged home on a slow steroid-tapering regime and continues to show clinical improvement at the time of writing. He has been established on monthly nebulised pentamidine for PJP prophylaxis for 6 months following cladribine therapy. There has been full recovery of blood counts 2–3 months later.
Discussion
Diagnosis and triggers
HLH is a rare diagnosis characterised by immune dysregulation and uncontrolled proliferation of lymphocytes and macrophages. These lymphocytes and macrophages secrete large volumes of inflammatory cytokines which cause the development of symptoms. 6 There are three salient diagnostic considerations:
Making a diagnosis of HLH: the combination of clinical and laboratory features, and the H-score was supportive of a diagnosis of HLH in our patient.
Distinguishing primary from secondary HLH: primary HLH is typically diagnosed in childhood and is uncommon in adults. 7 Primary HLH was unlikely in our 45-year-old patient.
Identifying the cause: this allows for treatment or removal of the trigger, reducing the risk of recurrence. In our case, the trigger was favoured to be cotrimoxazole.
Infectious triggers account for >50% of cases of secondary HLH. 8 Despite extensive investigations into our patient, no infectious agents were identified. It was still appropriate to treat using the neutropenic sepsis protocol as our patient was immunocompromised.
Co-trimoxazole-induced HLH was considered likely in our patient given the temporal association. As per local PJP prophylaxis policy, our patient was prescribed 960 mg co-trimoxazole three times per week (Monday, Wednesday and Friday), first administered 14 days prior to his presentation with fevers and rigours. Furthermore, the only instance prior to initiation of steroids that our patient defervesced was after presumed clearance of co-trimoxazole – this defervescence occurred on a Sunday with the prior dose being given on Friday morning (Figure 1(c)). Co-treatment with antibiotics and steroids made the initial identification of HLH challenging. Other case reports of co-trimoxazole-induced HLH also occurred within 12 days of starting treatment and showed resolution of pyrexia 48-h post-steroid therapy. 9 This timeline aligns very closely with the onset and clinical response in our patient.
There remain a number of alternative possible causes. In particular, several other medications are recognised as potential triggers of HLH.10,11 We considered cladribine and G-CSF as possible causes but the timeline did not align as closely as with co-trimoxazole. The half-life of cladribine is <20 hours, so the symptoms would have developed earlier if cladribine had been the trigger. 12 Furthermore, there are limited published data on cladribine triggering HLH. G-CSF was excluded due to symptoms starting prior to administration of G-CSF.
The history of hairy cell leukaemia was also pertinent and we assessed whether this malignancy was contributing as an HLH trigger. At diagnosis, >90% of the marrow cells represented hairy cell leukaemia using positive CD20, Cyclin D1 and DBA.44 on immunohistochemistry. On the repeat bone marrow, it was not possible to exclude the low-volume persistence of hairy cell leukaemia due to the diffuse phagocytic infiltrate (Figure 2(b)). Nonetheless, the repeat trephine showed a considerable reduction in disease burden with <1% of marrow cells being CD20 positive. It was not possible to repeat flow cytometry as the aspirate was relatively acellular.
Management
There are several aspects of managing HLH to consider:
1. Treatment of underlying triggers
In our case, we sought opinions from microbiologists and infectious diseases specialists; infection was felt unlikely after extensive investigation and this influenced our decision-making in stopping antimicrobials, including co-trimoxazole.
2. Supportive management
Our patient required extensive support with IV fluids for electrolyte abnormalities and blood products for DIC. Prophylaxis of opportunistic infection is also important and, as previously mentioned, nebulised pentamidine has been commenced monthly for PJP prophylaxis.
3. Immune suppression/HLH-directed treatment
Much of HLH management has been influenced by the pivotal HLH-94 13 and HLH-2004 14 studies. These trials were performed in selected cohorts of patients and involved combinations of high-dose dexamethasone with chemotherapeutic agents such as etoposide. Novel, targeted agents including anakinra 5 or alemtuzumab 15 are available for HLH.
Similar to another published case report of drug-induced HLH, 9 our patient responded rapidly to steroid monotherapy and did not require second-line therapies.
Learning points
▪ HLH is a rare inflammatory condition with fatal consequences if not diagnosed and treated rapidly. Its diagnosis requires recognition of HLH, identification of a trigger and distinguishing primary from secondary causes.
▪ Drug-induced HLH is an uncommon cause of secondary HLH. Co-trimoxazole-induced HLH is a very rare complication of a commonly used drug.
▪ Drug-induced HLH may respond to steroid monotherapy and drug cessation.
Footnotes
Authorship statement
KS and CR wrote the first draft and contributed equally to this case report. All authors contributed to the editing and writing of the final manuscript. All authors were involved in the management of the patient.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Written informed consent for the paper to be published was obtained from the patient for publication of this paper, including accompanying images.