
Abstract
The effectiveness of any healthcare-related research governance system is dependent on its ability to identify, challenge and change practices that compromise its ability to deliver timely, proportionate review. We present a case study outlining our experience of obtaining research ethics committee (REC) and Public Benefit and Privacy Panel (PBPP) for Health and Social Care approval to conduct a study which aimed to collect data on diagnostic and care pathways and determine the national prevalence of two rare diseases in Scotland. We discuss the threats posed to low-risk observational epidemiological research by disproportionate governance practices and propose practical solutions. In the context of increasing investment, the ever-increasing barriers to doing high-quality, low-risk epidemiological research using patient-identifiable information is concerning. Information governance committees, guided by clinical researchers, must step up as leaders in this area, making use of flexibilities and opportunities within the law.
Keywords
Background
The UK has positioned itself as an international leader in the use of routinely collected data to transform healthcare delivery and research, with significant financial buy in from national research councils and devolved governmental health and research departments. All healthcare-related research must adhere to regulations and standards to promote ethical and scientific quality and meet legal data protection requirements. Implementation of this governance framework results in a variety of approval procedures by several national and regional regulatory and monitoring bodies which must be successfully navigated before any research study can begin. The effectiveness of any governance system is dependent on its ability to identify, challenge and change practices that compromise its ability to deliver timely, proportionate review and elicit confidence and trust from both research participants and researchers. Drawing on our experience of navigating governance approval processes in Scotland, a country which due to its population characteristics and health service structure should be excelling in epidemiological and health data science research, we discuss the threats posed to high-quality, low-risk, observational epidemiological research by disproportionate governance practices.
Case study
We sought approval from both the research ethics committee (REC) and the Public Benefit and Privacy Panel (PBPP) for Health and Social Care to conduct a study, peer reviewed and funded by a Chief Scientist Office clinical research fellowship in progressive supranuclear palsy (PSP) and corticobasal degeneration which aimed to determine the national prevalence of these two rare and difficult to diagnose neurological diseases and collect data on diagnostic and care pathways. In order to determine prevalence, case ascertainment methods were rigorously designed to maximise the accuracy of the prevalence rate by avoiding both underestimation (requiring multiple, overlapping methods of case identification including clinician referral and the use of ICD-10 diagnostic codes from the Scottish Morbidity Record (SMR)) and overestimation (requiring identifiable information (name, Community Health Index (CHI), diagnosis) to deduplicate referrals and confirm diagnostic accuracy). In addition, to minimise selection bias relating to our diagnostic and care pathways objectives, we requested that identified prevalent cases were invited to participate in these additional objectives via invitation letter, signed and addressed by their local clinician but returned to the research team to send to patients, aiming to reduce clinician workload, avoid missing or duplicate invitations, and monitor non-response (proposed data flow detailed in Figure 1).
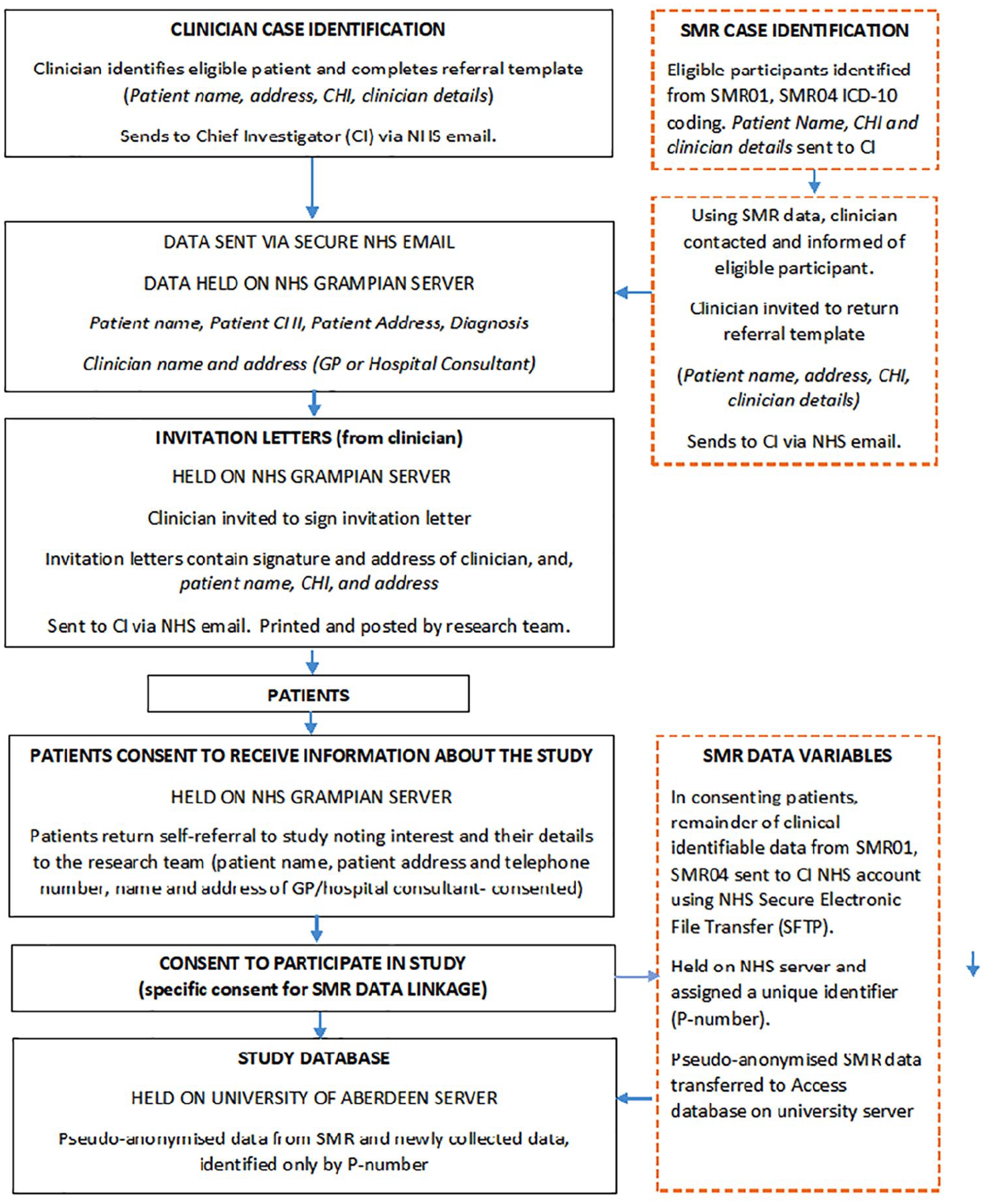
Proposed flow of data.
Due to sequentially cancelled and full meetings, it was nearly 3 months from submission until our application was initially reviewed by the REC. There were significant delays before our application was submitted by our electronic Data Research and Innovation Service (eDRIS) coordinator to the PBPP due to requests for additional applications (e.g., Privacy Impact Assessment) and in awaiting separate approval from the National Health Service Central Register. While at a Tier 1 level, the application to the PBPP was reviewed in a timely manner (20 days), in successive tiers of PBPP review (each resubmission coordinated by an eDRIS coordinator), time intervals became longer (Table 1). When our PBPP application had been reviewed by both the Tier 1 and Tier 2 panel, with written responses provided to both, and with a further Tier 2 response pending, we received an unfavourable REC opinion. Their primary objection related to the provision of unconsented patient data from clinicians and the SMR to the research team. The proposed solution to this objection was to separate the original proposal into two applications: a prevalence ‘audit’ (not necessitating REC review but requiring PBPP approval) permitting the proposed collection of unconsented patient-identifiable information to determine prevalence, and a research study incorporating our aims relating to diagnostic and care pathways (requiring both REC and PBPP approval), restricted to consented clinician referral or patient self-referral. At this stage we contacted the PBPP for advice. An eDRIS advisor relayed that it would be inadvisable to wait for the PBPP Tier 2 Committee formal opinion as the application would likely be rejected outright, ‘re-setting’ the application timeline to the beginning. We were advised to ‘pause’ the process and revise our response. A new full application separating the original proposal into the advised prevalence ‘audit’ and separate research study (now approved by the REC, approximately 6 months after initial submission) was submitted to the Tier 2 (Out of Committee), and subsequently escalated to the Tier 2 (Full Committee). After two further written responses to the Tier 2 Full Committee, including supporting analyses, further amendments and a telephone conversation between the study researchers and the chair of the PBPP panel, the amended application was approved, nearly a year after submission.
Timeline of application process.
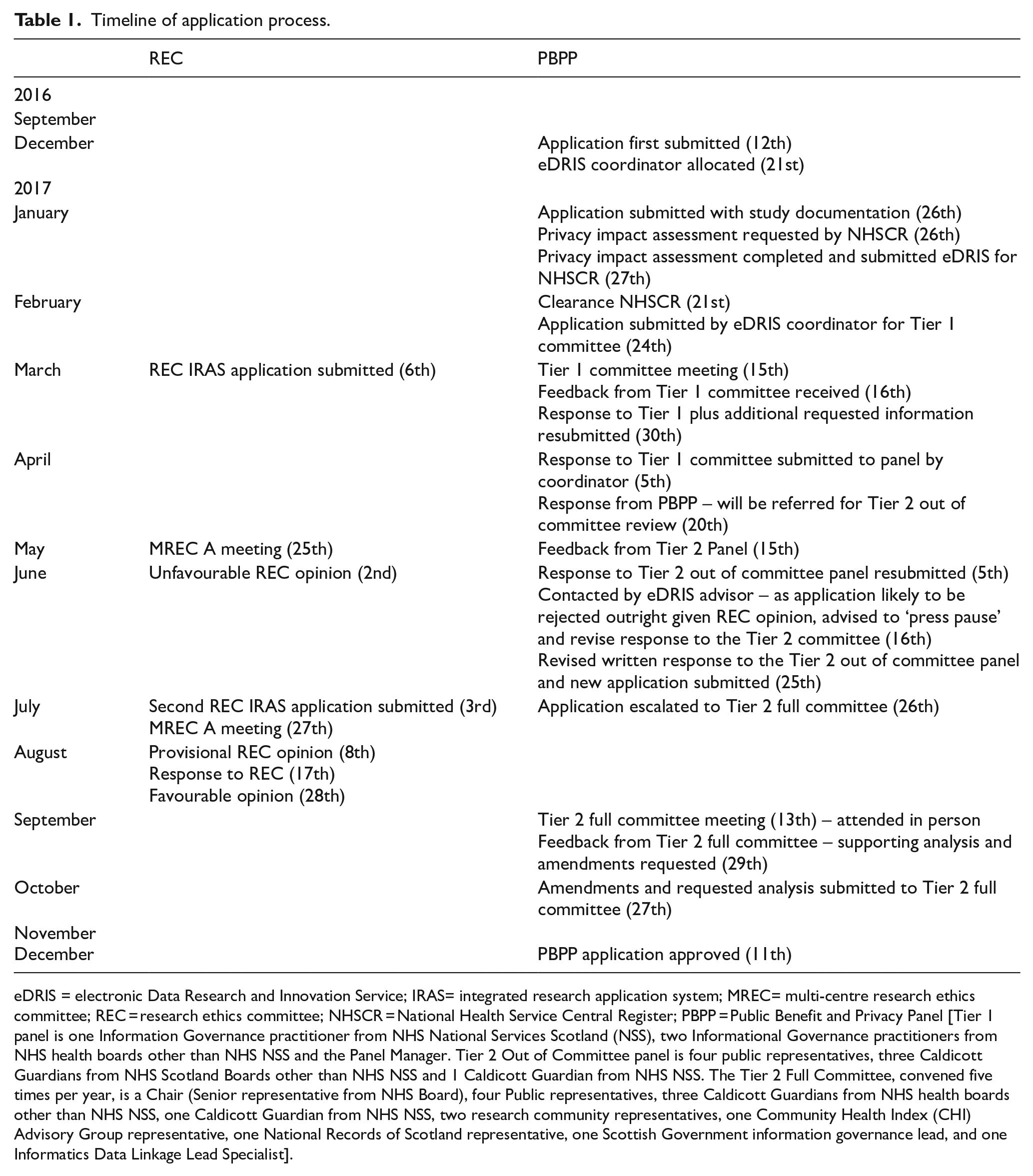
eDRIS = electronic Data Research and Innovation Service; IRAS= integrated research application system; MREC= multi-centre research ethics committee; REC = research ethics committee; NHSCR = National Health Service Central Register; PBPP = Public Benefit and Privacy Panel [Tier 1 panel is one Information Governance practitioner from NHS National Services Scotland (NSS), two Informational Governance practitioners from NHS health boards other than NHS NSS and the Panel Manager. Tier 2 Out of Committee panel is four public representatives, three Caldicott Guardians from NHS Scotland Boards other than NHS NSS and 1 Caldicott Guardian from NHS NSS. The Tier 2 Full Committee, convened five times per year, is a Chair (Senior representative from NHS Board), four Public representatives, three Caldicott Guardians from NHS health boards other than NHS NSS, one Caldicott Guardian from NHS NSS, two research community representatives, one Community Health Index (CHI) Advisory Group representative, one National Records of Scotland representative, one Scottish Government information governance lead, and one Informatics Data Linkage Lead Specialist].
Over the course of the approvals process various objections and changes to the original study design were proposed, each requiring a written response. The PBPP Tier 2 (Full Committee), for example, objected to transferring identifiable information outside individual health-boards, proposing identifiable information was collated and retained within individual health-boards by a single responsible clinician. This would have required increased resource (a named clinician in each health-board to monitor and de-duplicate patients identified by different sources) and would still have necessitated the eventual transfer of identifiable data outside individual health-boards to determine a national prevalence rate. The PBPP also sought justification for not using a data-safe haven for SMR data. The premise of the safe haven environment is that researchers are not permitted access to patient identifiers. If identifiable information were transferred as suggested, data would subsequently have needed to have been physically transferred out of the safe haven environment (to contact clinicians to confirm the SMR ICD-10 diagnoses) or brought into the safe haven (identifiable information received by clinician notification deduplicated against SMR identified cases), creating additional, unnecessary data risks. The use of a single clinician per health-board to coordinate notification of prevalent cases and issue invitation letters relating to diagnosis and care was also advised, but was felt to pose an unreasonable burden on clinicians, likely reducing clinician engagement, skewing the participation of health-boards and restricting the number and geographical spread of referred patients.
An appreciated risk in the original design was the breach of privacy resulting from the unconsented release of identifiable information to the researchers (who were NHS clinicians) for the purposes of determining prevalence. For objectives relating to diagnosis and care, whilst the simplest way to address these was to ask prevalent patients if they wished to give informed consent to partake in these study objectives, an inherent risk was that researchers could use identifiable information provided to determine prevalence to gather unconsented clinical information. However, we felt the proposed alternatives were impractical, threatened the scientific validity of the prevalence study, and had little impact on data security, and so we considered the potential harms were necessary to answer the specific question posed, were for legitimate medical purposes and proportionate to the broader public benefit/gains. Measures to safeguard personally identifiable clinical information, including requesting the minimum necessary unconsented or pre-consented sensitive information, restricting access to the chief investigator and data custodian (both NHS clinicians fully aware of the responsibilities and duties of respecting patient confidentiality), and employing safe data storage and transfer methods as advised by a professional data management team, were employed. No patient would have been directly contacted by the research team without their explicit permission.
Reflections on the approvals process
The decision of the REC to redefine a prevalence study as audit is difficult to reconcile. Prevalence and incidence studies, especially high-quality studies, acquire new data and are not measuring performance against a standard, and are, therefore, research, not audit. By effectively prohibiting the undertaking of active full case ascertainment, large high-quality prevalence and incidence studies become impossible. Beyond playing ‘hot potato’ with potentially contentious or sensitive decisions, the lack of clarity of the respective roles of each governance body results in unnecessary duplication of reviews, revealing inconsistency in decision making. For example, non-substantive changes to language within study documentation approved by both the sponsor and REC was required by the PBPP, necessitating the submission of amendments to both the sponsor and REC to meet their respective legal and ethical requirements. In parallel, the PBPP appeared unwilling to provide an independent decision on the use of patient-identifiable information, despite such decisions falling within their governance remit (‘. . .many of the panel’s concerns would be allayed by an OK from the ethics committee. . . it might be a good idea to discuss these issues with them before coming again to PBPP. . .’). This lack of agreement or transparency in the definitions of research or audit, and where the boundaries between the respective roles and responsibilities of the REC and PBPP lie, is extremely problematic.
Governance review should be rigorous, efficient, and with predictable timelines. This is essential given the time-limited nature of grants and because most researchers apply only when funding is secured. The structure of the PBPP review process creates numerous opportunities for delay. While the assistance of the eDRIS team is well-intentioned, the reality of such gatekeeping measures is an unrestricted (and presumably unaudited) period before an application is submitted for review. As reviews are escalated, additional delays are introduced due to the infrequency of meetings. Providing written responses and additional information to each panel tier also involves significant opportunity cost and when requested information has been provided, or when requests become repetitive, insubstantial or with little evidence previous responses have been acknowledged, the value of such requests becomes doubtful. Requested information from higher tiers of the PBPP seemed, at times, unnecessarily authoritarian: ‘. . .Please provide minutes of the review of the application by the PSP Association, which has agreed to support the application. . .’, or, ‘. . .any requests for sharing of data with other researchers for other projects to be based on further PBPP approval of those projects. . .’ (despite explicit written consent being sought from participants to share their data with other bona fide researchers). Additional requests to engage R&D patient/public involvement leads more actively and undertake wider public engagement and consultation, made at late stages of the review process, threatened to entirely derail the ability to carry out the study within clinical research fellowship time. While patient/public involvement is crucial in research design and conduct (and now required by most funders), we do not believe they should be part of the remit of the PBPP review. Other requests such as – ‘. . .provide justification and explain the validity for broad consent to data sharing for future research. . .’, were perplexing as this is in stark contrast to the data-sharing expectations of many research councils and funders.
Good communication is essential, particularly when unexpected barriers or time-delays arise. There appeared to be a greater unwillingness within the PBPP to engage with researchers to advise upon and resolve identified issues. In the context of increasingly critical time delays, combined with information relayed through unofficial channels and explicit statements on official websites such as ‘. . .the determination of the committee is final. . .’, this reluctance to communicate created an uncomfortable feeling of powerlessness, relative to the ability of panel to reject or delay (without apparent limits) an application, irrespective of secured charity and government funding or ethical approval. In contrast, while we disagree with the decision of the REC to classify a prevalence study as audit, they were keen to facilitate the approval of the amended study and readily responded to emails requesting further feedback, advised on the most efficient way to navigate resubmission and approval, and nominated a specific individual on the REC to ensure the second application was approved in a reasonably timely manner. This conveyed an awareness of the detrimental impact of approval delays and an appreciation of the capacity within their role to either hinder or enable researchers. Unfortunately, the formed impression of the PBPP was of an unnecessarily obstructive, inefficient (or excessively overworked) and disproportionately risk averse governance body, who, by affording little professionalism or trustworthiness to their applicants, inadvertently give the impression that all researchers start from a premise of doing harm, which is extremely demoralising.
Solutions (see Key points)
Clear standardised governance pathways, specifically for incidence and prevalence studies, must be agreed upon between the REC and PBPP. This is especially important for rare diseases where national or regional studies, rather than local studies, are essential to ensure robust sample sizes. Access to a limited amount of unconsented routinely collected data is required to maximise scientific quality. This unconsented clinical information is often not suitable or appropriate for data safe havens because of the need to check clinical diagnoses, particularly important in difficult-to-diagnose conditions. There must also be an understanding that such epidemiological studies are research and not audit and, to avoid a disproportionate response, that they are low risk. Explicit agreement on these measures is essential to ensure future researchers do not unnecessarily revisit the same issues we faced.
A clearer practical distinction of the roles and responsibilities of the REC and PBPP is also required. We believe the number of tiers within the PBPP approval structure should be reconsidered. At a minimum, proposals escalated beyond a Tier 1 review should be flagged so that proactive steps are taken to facilitate communication with applicants. If the PBPP continue to require operational and administrative support by eDRIS, a time-limit should be imposed on this initial preparatory process. Requests for clarification or additional information, particularly in the context of a review by another approval body, should add value and be solely to assist panels to resolve governance difficulties; that is facilitate their ability to identify risks posed by the research protocol, determine if risks are disproportionate to potential research benefits or if the study can be redesigned to reduce risk without compromising its ability to answer the research question, and finally, ensure investigators have taken reasonable steps to minimise (note, not necessarily eliminate) the chance that remaining risk results in harm. Regional and national committees should also aim to provide greater consistency to have greater legitimacy in the eyes of the research community and the public.
Conclusions
In the context of increasing investment, the simultaneous, ever-increasing, barriers to doing important, high-quality, low-risk epidemiological or population-based research using patient-identifiable information is concerning. At best, this runs the risk of making such research poorer quality. At worst, it may kill it. Continuing to tolerate excessive and unnecessary bureaucracy will serve the interests of neither the public nor research community. The historical impetus for research governance arose, in part, due to concern relating to increasing commercial research and the necessity to both facilitate and monitor this involvement to protect both patient and NHS interests. Ironically, if approval processes continue to be so prohibitively time-consuming, high-quality, low-risk epidemiological research will become possible only by industry or by universities with an extensive research infrastructure and dedicated administrators. When, in the interests of upholding absolute privacy at all costs, we do not do what would have most benefited the patient/public’s best interests, and where money from charities and other research funders goes into navigating needless bureaucracy rather than carrying out research, patients and funders will lose confidence, to the detriment of evidence-based clinical care or public health provision. Information governance committees, guided by clinical researchers, must step up as leaders in this area, making use of flexibilities and opportunities within the law to enable present and future researchers to maximise the benefits of routinely collected data.
Key points
The current approvals process for low-risk epidemiological research in Scotland is prohibitively time-consuming and bureaucratic, duplicates aspects of review and is disproportionately risk averse. This threatens future research.
Access to a limited amount of unconsented routinely collected data is required in these studies to maximise scientific quality.
This unconsented clinical information is not suitable for data-safe havens because of the need to check clinical diagnoses.
The current hierarchical structure of the Public Benefit and Privacy Panel (PBPP) submission and review process creates numerous opportunities for duplication and delay.
Explicit agreement that prevalence and incidence studies are research, and not audit.
Develop standardised governance pathways, specifically for incidence and prevalence studies.
These pathways must make clear and practical distinctions on the respective roles and responsibilities of the research ethics committee (REC) and PBPP.
Requests from governance bodies for additional information should be limited to their defined remit.
Ensure investigators have taken reasonable steps to minimise (note, not necessarily eliminate) risk.
Review role and structure of PBPP.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr Swallow received a clinical research fellowship jointly funded by the Chief Scientist Office (CSO) of the Scottish Government and PSP Association.