
Abstract
Introduction
Lumbar disc herniation (LDH) involves inflammation and metabolic stress. Tissue-specific renin-angiotensin-aldosterone system (RAAS) signaling may act as an upstream regulator of disc degeneration. This study investigates RAAS dysregulation as a central driver of LDH and its contribution to inflammation, metabolic reprogramming, and disc injury.
Methods
Bulk and single-cell transcriptomics assessed RAAS activity, while MR/GWAS analyses examined causality. Machine learning with the MIMIC database identified biomarkers. In vitro nucleus pulposus cell experiments validated the ACE2/Ang(1–7)/MAS1 protective axis.
Results
RAAS components ACE, AGTR1, ACE2, AGTR2, and MAS1 were dysregulated in LDH across disc cells, immune populations, and endothelium, with a shift toward glycolysis. Patients showed elevated Ang II/Ang(1–7) ratios. Ang II induced NF-κB mediated inflammation, extracellular matrix degradation, and apoptosis, reversible by losartan or protective axis activation.
Conclusion
RAAS dysregulation serves as an upstream hub driving inflammation, metabolic imbalance, extracellular matrix breakdown, and disc cell injury in LDH. Therapeutic strategies should suppress the ACE/Ang II/AT1 axis while enhancing the ACE2/Ang(1–7)/MAS1 protective pathway.
Integrated multi-omics analyses identify renin–angiotensin system dysregulation as a key driver of lumbar disc herniation. Classical ACE/Ang II signaling promotes inflammation and matrix degradation, whereas the ACE2/Ang-(1–7)/MAS1 axis counteracts degenerative processes.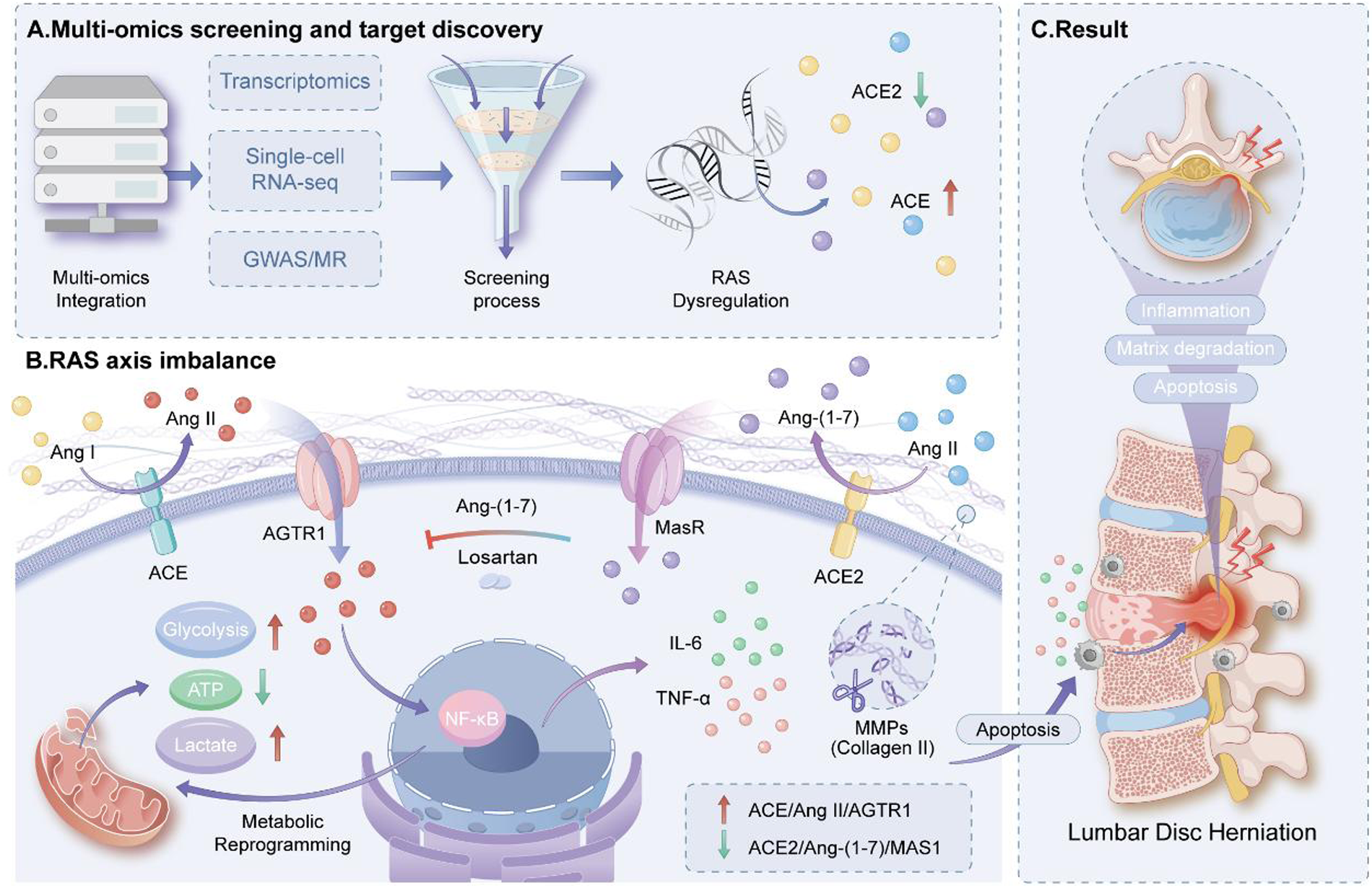
Keywords
Get full access to this article
View all access options for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.