
Abstract
Introduction
The study of the receptors of the renin-angiotensin and kinin-kallikrein systems and their interactions has enhanced our understanding of hypertension physiology and contributed to the development of improved pharmacological treatments (McCarthy et al. 2025).
Objective
This work aimed to investigate the functional interactions among AT1, AT2, and B2 receptors specifically the effects of agonist-agonist, agonist-antagonist, and antagonist-antagonist combinations on vasocontractile response to Ang II.
Methods
Concentration-response curves to Ang II were performed in WKY and SHR rat aortic rings in the presence of bradykinin (BK), valsartan, CGP42112A, HOE140, and their combinations. NO levels were indirectly determinated by the Griess reaction in aortic rings.
Results
Ang II induced a stronger contractile response in SHR compared with WKY. AT1R antagonism and AT2R receptor stimulation significantly reduced vasoconstriction and increased NO levels. BK also attenuated the contractile response without increasing NO levels. In contrast, blockade of B2R or AT2R did not reduce maximal contraction. Combined treatments involving valsartan or CGP42112A decreased Ang II reactivity.
Conclusion
Activation of AT2R and B2R counteracts Ang II–induced vasoconstriction through NO-dependent and NO-independent mechanisms. Receptor heterodimerization appears to contribute to these modulatory effects.
Introduction
Hypertension is a cardiovascular disease that has a high incidence, prevalence, and mortality worldwide. 1 This condition is associated with endothelial dysfunction, where relaxing factors such as nitric oxide (NO), endothelium-derived hyperpolarizing factor (EDHF), and prostacyclin (PGI) are reduced, while vasoconstrictor factors such as endothelin, catecholamine, and angiotensin II (Ang II) are increased. The renin-angiotensin system (RAS) was discovered 125 years ago and was first described as a pressor agent in hypertension and ischemic renal disease, since then extensive research has revealed its crucial role in regulating hemodynamic homeostasis and how its dysfunction contributes to the development of hypertension and various vascular alterations. 2 The main effector of RAS is Ang II, which acts on two receptors subtypes: AT1 and AT2. Although AT2 shares 42% of homology with AT1, it exerts the opposing effects. However, our understanding of the AT2 receptor in hypertension is still evolving.3,4 Another system that generates vasoactive peptides from plasma can be traced back to the nineteenth century when a urinary substance named kallikrein was discovered. 5 It was later found that kallikrein itself is not vasoactive but instead releases biologically active peptides, known as kinins, from plasma protein. Bradykinin, named after the slow contraction it induces in guinea pig ileum, acts through two receptors, B1 and B2. The connection between these two systems lies in the Angiotensin-Converting Enzyme (ACE), which metabolizes biologically active peptides from both the kallikrein-kinin system and the RAS. 6
The AT2 and B2 receptors regulate vasodilatory functions, by promoting NO production. In hypertension, vasodilator mechanisms associated with B2 receptors are also impaired. Both angiotensin and bradykinin receptors belong to the family of G protein coupled receptors (GPCRs), which play key roles in numerous physiological and pathological processes.7,8 These receptors act as intracellular signal transducers, translating ligand binding into cellular responses. Studies have shown that GPCRs can form homo- or heterodimers which may alter physiological, pathophysiological, and pharmacological outcomes. 9
Heterodimerization of GPCRs between RAS and bradykinin receptors can significantly modify G-protein coupling and downstream signaling, thereby contributing to hypertension. AT1/B2 dimers enhance Ang II responsiveness, activate Gαq/11, increase endothelin-1 release, and are associated with preeclampsia. AT2/B2 dimers modulate nitric oxide production and kinase/phosphatase pathways, influencing cardiovascular and renal function. 10 The physical association among these types activates multiple crosstalk signaling pathways, inhancing NO and cGMP production. 7 These receptor interactions represent promising targets for the development of novel antihypertensive therapies, as current pharmacological treatments often require the combination of two or more drugs to achieve th optimal control. Therefore, further research is needed to clarify the functional interactions between AT1, AT2, or B2 receptors in the vasodilator and vasoconstrictor responses induced by Ang II. This study may open a window to the possibility of testing potential drug combinations. The objective of the present study whether combinations of AT1, AT2 or B2 receptor agonists and antagonists functionally modify the vascular response to Ang II and the NO levels in the aorta of spontaneously hypertensive rats.
Methods
Isolated organ bath experiment
Concentration–response curves (CRC) to angiotensin II (Ang II; 1 × 10−10–1 × 10−6 M) were generated in the absence or presence of the following compounds: bradykinin (1 × 10−5 M), CGP42112A (1 × 10−5 M), valsartan (1 × 10−9 M), PD123319 (1 × 10−5 M), HOE140 (1 × 10−5 M), or their combinations:
CGP42112A + bradykinin (both 1 × 10−5 M) CGP42112A + HOE140 (both 1 × 10−5 M) valsartan + bradykinin (1 × 10−9 and 1 × 10−5 M, respectively) valsartan + CGP42112A (1 × 10−9 and 1 × 10−5 M, respectively) valsartan + PD123319 (1 × 10−9 and 1 × 10−5 M) valsartan + HOE140 (1 × 10−9 and 1 × 10−5 M).
Each compound (alone or in combination) was incubated for 15 min before constructing the Ang II CRC. After completion of the experiments, aortic rings were collected for nitric oxide (NO) quantification.
NO indirect measurement. NO levels were determined using Griess reaction in thoracic aortic rings collected after the organ bath experiments. This method measures nitrite, a stable NO metabolite, spectrophotometrically. First, nitrates were reduced to nitrites using cadmium-cupper granules. Then nitrites reacted with sulfanilamide (1%) to form a diazonium compound, which subsequently coupled with N-ethylenediamine (0.1%) (reagents from Merckgroup Mexico) to generate an azo-chromophore compound that absorbs at λ=540 nm. It should be noted that NO quantification is not direct and only represents 75% of the actual concentration. The Griess technique measures the amount of nitrites present in the samples.
Statistical analysis. Data from vascular reactivity experiments were expressed as mean ± SEM from 12 aortic rings from six rats per group. NO levels were expressed as mean ± SEM from six aortic rings, each measured in triplicate. EC50 values were obtained from the Ang II CRC for each ring using a non-linear regression using the Hill equation, and the mean ± SEM was calculated for each group. Statistical differences were assessed by one-way or two-way ANOVA followed by Tukey's post hoc test. A p value <0.05 was considered statistically significant.
Results
The contractile response to Ang II in aortic rings from SHR is shown in Figure 1. The contraction was significantly greater in aortic rings from SHR than WKY confirming an enhanced vascular reactivity in hypertension.
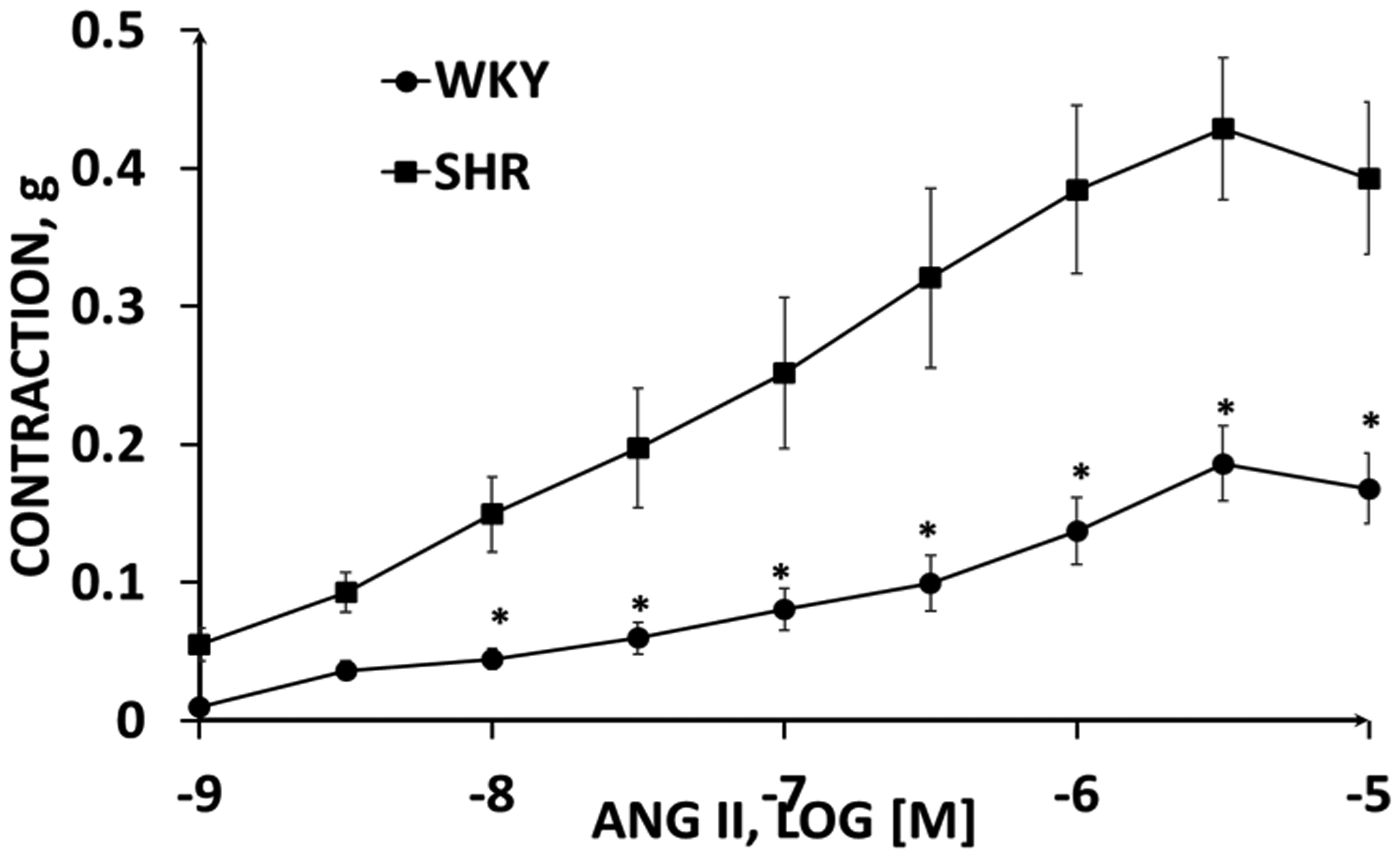
Concentration response curves to Ang II using aortic rings with endothelium from Wistar Kyoto and SHR rats. Results are expressed as mean ± SEM of 12 aortic rings of 4 rats. Two-way ANOVA followed by a post hoc Student-Newman Keuls test *P < 0.05 vs SHR.
Figure 2 shows the vasocontractile response to Ang II in the presence of agonists and antagonists of AT2R, B2R, and AT1R: CGP42112A (AT2R agonist), PD123319 (AT2R antagonist), BK (B2R agonist), HOE140 (B2R antagonist), and valsartan (AT1R antagonist). Activation or blockade of AT1R, AT2R, and B2R receptors significantly modulated this response. Most receptor agonists and antagonists attenuated Ang II-induced vasoconstriction, except for the B2R antagonist HOE140, which had no significant effect. In table 1, EC50 of the curves are shown.

Concentration response curves to Ang II using aortic rings with endothelium from SHR rats in absence or in presence (10-7 M) of CGP42112A an AT2 agonist, PD123319 and AT2 antagonist, valsartan an AT1 antagonist, bradykinin (BK) an B2 agonist, HOE140 and B2 antagonist. Results are expressed as mean ± SEM of 12 aortic rings of 4 rats. Two-way ANOVA followed by a post hoc Student-Newman Keuls test *P < 0.05 vs SHR.
CE50 values (µM) for Ang II in SHR.

Results are expressed as mean ± SEM of 12 aortic rings of 4 rats. Two-way ANOVA followed by a post hoc Student-Newman Keuls *P < 0.05 vs WK; aP < 0.05 vs SHR and bP < 0.05 vs the same group without combination.
Figure 3 shows the vasocontractile response to Ang II in the presence of AT2R agonist (CGP42112A) combined with either the B2R agonist (BK), the B2R antagonist (HOE140), or the AT1R antagonist (valsartan). Combined stimulation of AT2R and B2R, or inhibition of AT1R, further reduced Ang II-induced contraction, suggesting functional cross-talk between these receptor systems (Table 1).

Concentration response curves to Ang II using aortic rings with endothelium from SHR rats in absence or in presence of combination: CGP42112A with bradykinin (BK), HOE140, or valsartan. Results are expressed as mean ± SEM of 12 aortic rings of 4 rats. Two-way ANOVA followed by a post hoc Student-Newman Keuls test *P < 0.05 vs SHR.
Figure 4 shows the vasocontractile response to Ang II in the presence of AT1R antagonist (Valsartan) combined with B2R antagonist (HOE140), AT2R agonist (CGP4211A), B2R agonist (BK) or AT2R antagonist (PD123319). The combinations of valsartan with CGP42112A, BK or PD27319 significantly reduced Ang-II-induced vasoconstriction compared with the other combinations. Interestingly, valsartan (AT1R antagonist) markedly decreased the contractile response, but its effect was reversed when co-administered with HOE140, indicating that B2R activity contributes to the vasodilatory influence of AT1R blockade.

Concentration response curves to Ang II using aortic rings with endothelium from SHR rats in absence or in presence of combinations: Valsartan with HOE140. CGP42112A, bradykinin (BK), or PD123319. Results are expressed as mean ± SEM of 12 aortic rings of 4 rats. Two-way ANOVA followed by a post hoc Student-Newman Keuls test *P < 0.05 vs SHR.
Figure 5 shows NO levels in the presence or absence of AT1R antagonist, B2R agonist or antagonist, and AT2R agonist or antagonist allone or in combination in aortic rings from SHR and normotensive rats. Nitric oxide (NO) production was elevated in the presence of CGP42112A (AT2R agonist) and valsartan, consistent with their vasorelaxant effects, whereas BK, PD123319, and the CGP42112A + BK combination were associated with lower NO levels.

No quantification. These results are the mean ± SEM (n = 6). Two-way ANOVA followed by a post hoc Student-Newman Keuls test, all groups were different from WK; *p < 0.05 vs SHR (Veh). Another significant difference was found BK vs Valsartan (P = 0.0001); CGP42112A vs Valsartan (P = 0.032); PD123319 vs Valsartan + PD123319 (P = 0.004); Valsartan vs Valsartan + BK (P = 0.001); Valsartan + BK vs Valsartan + PD 123319 (P = 0.04).
Discussion
Essential hypertension is a multifactorial pathology of unknown etiology, characterized by endothelial dysfunction, particularly involving functional imbalances between the vasoconstrictor and vasodilator endogenous substances related to the RAS and KKS. In this study we examined the interactions among AT1, AT2 and B2 receptors in the pressor response to Ang II, since AT2 and B2 receptors has been described as counter-regulatory axis to AT1 receptor ativity. The present work provides new insights into the mechanisms underlying Ang II–induced vascular reactivity in hypertension. We demonstrate that activation of AT2R or B2R attenuates vasoconstriction in aortic rings from SHR rats. Moreover, AT1R antagonism and AT2R stimulation enhanced NO production, while blockade of AT2R or B2R failed to reduce maximal contraction, highlighting the complexity of receptor interactions. Together, these results highlight a dynamic interplay between the AT1R, AT2R, and B2R pathways, where AT2R and B2R activation counteracts Ang II–mediated vasoconstriction, partly through enhanced NO bioavailability. This supports the concept that modulating AT2R and B2R signaling may help restore vascular balance in hypertension.
The Ang II is the main peptide of RAS, binds with the high affinity to the AT1 and AT2 receptors. 11 Ang II interaction with AT1R coupled to Gq proteins, activates phospholipase C, leading to the formation of -inositol 14,5-triphosphate (IP3). This signaling cascade induces a contractile response through Ca2+ release from intracellular stores in vascular smooth muscle cells. 20 Conversely, blocking AT1R inhibits Ca + 2 release, promoting vasodilation 12 . In our results, valsartan significantly reduced the Ang II-induced contraction in SHR rats, accompanied by increased NO levels. Consistent with previous studies Ang II or its metabolites can stimulate NO and cGMP production through AT1R antagonism with valsartan.13,14 It has been suggested that this effect results as a concurrent stimulation of AT2R and B2R 15 ; however, when both receptors are blocked, the AT1R antagonist effect on NO production is abolished. 16 Thus, NO production appears to depend on receptor interactions involving agonists and antagonists of AT1R, AT2R and B2R. 17
Activation of AT2R by Ang II contributes beneficially by modulating the vasoconstrictor effects mediated by AT1R stimulation. Although AT2R is considered an endogenous physiological antagonist of AT1R. However, the underlying mechanisms remain unclear, and some evidence suggests AT2R may mimic AT1 receptor functions under specific conditions. 18 Therefore, selective agonists and antagonists have been developed to better define the physiological or pathophysiological roles of AT2R. In this context, CGP42112A a selective AT2R agonist that has been instrumental in elucidating cardiovascular signal pathways. 19 Activation of AT2R by CGP42112A in vascular smooth muscle cells enhances NO production through the catalytic action of eNOS, leading the activation of soluble guanylyl cyclase and increased cGMP forming the AT2R-eNOS-NO axis responsible for vasodilation 20 . In the present study AT2R stimulation with CGP42112A reduced Ang II-induced vascular reactivity and increased NO production.
Sanja Bosnyak et al. demonstrated that AT2R stimulation causes vasodilation in aortic and mesenteric arteries from normotensive and hypertensive rats and in aortic rings in vitro, though it does not necessarily reduce blood pressure in vivo. 21 CGP42112A has been shown to lower blood pressure, an effect blocked by the AT2R antagonist PD123319. 22 Several mechanisms counteracting AT1R-mediated vasoconstriction via AT2R activation has been described. 19 In our work AT2R activation with CGP42112A reduced the Ang II-induced contraction, reducing Ang II potency. These findings indicate that AT2R antagonism may interfere with AT1R-Ang II interaction efficacy. Evidence suggests that AT1R and AT2R form heterodimers independently of Ang II with AT2R acting as a physiological antagonist of AT1R function. Understanding the role of these heterodimers is crucial in cardiovascular disease. 23
In our study, AT2R activation by CGP42112A increased NO levels, while PD123319 reduced them, suggesting that NO production varies depending on receptor activation or blockade in the signaling pathways engaged.18,22 AT1R and AT2R activation also appear necessary to induce a conformational changes leading to heterodimerization, which promotes AT2R internalization; a process inhibited by AT1R antagonism. 24
Previous in vitro studies in rat uterine artery using losartan and CGP42112 demonstrated reduced Ang II-induced constriction, 25 which was blocked by PD123319, consistent with our findings. However, combined valsartan-CGP42112 did not alter NO levels, suggesting that vasorelaxation involves NO independent mechanisms. Some studies have reported that simultaneous AT1R and AT2R blockade increases Ang (1-7) formation via ECA2, promoting an increase in NO synthesis. 26 In our results, valsartan-PD123319 combination did not alter NO levels. Miura S et al., 2010 proposed that AT2R-mediated inhibition of AT1R signaling occurs mainly via negative crosstalk at the PLC-β3 phosphorylation level, rather than through surface heterodimerization. 27
The B2 receptor is constitutively expressed in cardiovascular tissues and plays a central role in blood pressure homeostasis. Coupled to and Gi₂α, Gi₃α and Gq /11 proteins its activation triggers PLC mediated synthesis of IP3 and DAG. IP3 induces Ca2+ release form the sarcoplasmic reticulum, activating Ca2+/Calmodulin dependent kinase, which promotes eNOS/NO/GCs/cGMP/PKG signaling, leading to vasodilation. Alternatively DAG activates PKC which stimulates PLA2 and arachidonic acid synthesis, leading to prostacyclin production. This prostaglandin, via its GPCR, increases cAMP and inhibits PKA, blocking L-type calcium channels and causing smooth muscle relaxation. 28 BK can also induce endothelium-dependent relaxation through NO release or contraction via endothelial B2R and prostaglandin H2. 25
In our study, BK reduced Ang II-induced contraction, likely through one or more of mechanisms mentioned above. The vasodilatory response mediated by B2R and AT2R activation appears to involve the AT2/B2/eNOS/NO axis. HOE140, a selective B2R antagonist is commonly used to explore this modulation. 29 In our experiments, HOE140 did not modify Ang II-induced contraction or NO levels in SHR aortic rings, suggesting the involvement of NO- independent mechanisms. Since bradykinin reduced Ang II induced vasoconstriction, B2R activation may counteract Ang II-mediated effects
Physical interactions between receptors such as AT1R-B2R heterodimerization, have also been described and may have therapeutic potential in cardiovascular diseases. AbdAlla et al. reported AT1R-B2R heterodimerization in rats and preeclamptic patients correlated with increased in B2R expression and hyperreactivity. In our combined AT1R and B2R antagonism did not reduce Ang II-induced contraction or change NO levels, 25 whereas AT1R antagonism combined with B2R agonism decreased contraction without affecting NO, indicating that B2R participation is essential for the vasodilatory effect of AT1R blockade. Transgenic mice lacking the AT1R-B2R heterodimers show a decreased vasopressor responses to AT1R activation 30 underscoring the importance of B2R in cardiovascular homeostasis.
Activation of AT2R and B2R counteracts Ang II-induced vasoconstriction, generating a vasodilator response. However, data regarding NO modulation and receptor regulation by the antagonists and agonists of AT2R-B2R receptors, particularly under hypertensive conditions, remain limited. The physical interaction between AT2R-B2R has been proposed to mediate modulatory effects in hypertension. Abadir et al. demostrated AT2-B2 heterodimerization in PC12 W cells, 7 although the physiological interactions is still unclear. In the present study, combined incubation with AT2R, B2R agonist or antagonist reduced Ang II-induced vasoconstriction. Further studies are required to elucidate the mechanisms involved. . Interestingly, Abadir, et al., also reported no significant increase in NO levels under similar conditions. Considering that SHR rats exhibit endothelial dysfunction, reduced NO bioavailability may explain these findings. 31
Finally, making a brief comparison between our findings and those found in females regarding the RAS, some results show that the Mas receptor (MasR) influences renal hemodynamics in a sex-dependent manner, with stronger effects in females. 29 When AT1R and AT2R are blocked, MasR enhances pressure natriuresis and diuresis in males but not in females. 32 Another group showed that Ang II produces greater renal effects in males, but these differences disappear after MasR blockade, suggesting that MasR modulates sex-specific vascular responses. In the 2K1C model, Ang II infusion caused dose-dependent increases in mean arterial pressure (MAP), while MasR blockade exaggerated renal vasoconstriction in ischemia–reperfusion (IR) and preconditioned IR (IPC + IR) groups. 33 Overall, MasR appears to have a protective, regulatory role in renal vascular function, mitigating Ang II–induced vasoconstriction, particularly under pathophysiological conditions. 34 These findings showed that RAS activity tends to show differences between sexes.
Conclusion
This study demonstrates that AT2R and B2R activation attenuates Ang II–induced vasoconstriction through both eNOS/NO-dependent and NO-independent pathways. The modulatory role of AT2R over AT1R function, together with the contribution of B2R, suggests that receptor crosstalk and potential heterodimerization are key mechanisms in vascular regulation. These findings highlight the importance of AT1R–AT2R–B2R interactions in essential hypertension and support their potential as therapeutic targets.
Limitations of the study
This study present some limitations such as the lack of in vivo blood pressure data. NO indirect measurements were restricted to ex vivo conditions. Also, the quantification of NO is indirect since, when using the Griess technique, nitrites are quantified, which represent 70% of the actual NO levels. A major limitation of this study is the lack of female participants; this study is necessary to determine if the observed effects differ between sexes.
Footnotes
Ethical statement
The care and use of the animals were conducted in strict accordance with the Guide for the Care and Use of Laboratory Animals (8th edition, National Academies Press). All procedures described herein were approved by the Internal Bioethics Committee of our institution (FES −01/16-05-2017) and complied with the Mexican Official Standard (NOM-062-ZOO-1999), which sets the technical specifications for the production, care, and use of laboratory animals. This project was carried out under the authorization of the Internal Committee for the Care of Experimental Animals of FES Cuautitlán (C18_04).
Author's contribution
Jazmin Flores and Luis Rocha, constructed and designed the manuscript. Ignacio Valencia wrote the original draft. Diego Lezama reviewed the manuscript. Diana Ramirez contributed to the revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: These study was supported by grants from DGAPA-UNAM PAPIIT IA204924, UNAM-PAPIIT IN204925, FESC CI-2429, PASPA Scholarship (Jazmín Flores-Monroy)
Dirección General de Asuntos del Personal Académico, Universidad Nacional Autónoma de México, Facultad de Estudios Superiores Cuautitlan, (grant number IA204924, IN204925, PASPA Scholarship, C2402, C2429).
Declaration of conflicting interest
The authors declare that this review was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data accessibility statement
All data are available from the corresponding author upon reasonable request.