
Abstract
Introduction:
An excess of angiotensin II (Ang II) causes hypertension and vascular injury. Activation of mitogen-activated protein kinase p38 (p38-MAPK) plays a substantial role in Ang II-dependent organ damage. Recently, we showed that p38-MAPK activation regulates the pressor response to Ang II. This study evaluates the effect of chronic p38-MAPK inhibition in Ang II-dependent hypertension.
Materials and methods:
C57Bl/6J mice were infused with Ang II for 14 days and either treated with the p38-MAPK inhibitor BIRB796 (50 mg/kg/day) or the vehicle as the control. We assessed vascular function in the aorta and isolated perfused kidneys.
Results:
Chronic p38-MAPK inhibition did not alter blood pressure at the baseline, but attenuated Ang II-induced hypertension significantly (baseline: 122 ± 2 versus 119 ± 4 mmHg; Ang II: 173 ± 3 versus 155 ± 3 mmHg; p < 0.001). In addition, BIRB796 treatment improved vascular remodeling by reducing the aortic media-to-lumen ratio and decreasing the expression of the membrane metalloproteinases (MMP) MMP-1 and MMP-9. Moreover, renal vascular dysfunction induced by chronic Ang II infusion was significantly ameliorated in the BIRP796-treated mice. Acute p38-MAPK inhibition also improved vascular function in the aorta and kidneys of Ang II-treated mice, highlighting the important role of p38-MAPK activation in the pathogenesis of vascular dysfunction.
Conclusions:
Our findings indicated there is an important role for p38-MAPK in regulating blood pressure and vascular injury, and highlighted its potential as a pharmaceutical target.
Keywords
Introduction
During the past decades, control rates and treatment options for hypertension have improved; however, hypertension is still the leading risk factor of mortality and morbidity in the world. 1 The need for better pharmaceutical treatment options remains high.2,3 Understanding the mechanisms which lead to hypertension and vascular dysfunction may contribute to the development of new potential targets for drug therapy.
Previously, we and others have shown that p38 mitogen-activated protein kinase (MAPK) is involved in myogenic control of the vascular wall in resistance vessels. 4 In addition, angiotensin II (Ang II)-dependent p38 MAPK activation is involved in vascular smooth muscle cell contraction via p38 MAPK-dependent phosphorylation of the myosin light chain (MLC20).5–9 Besides increases in contractile response, Ang II-dependent hypertension promotes changes in vascular wall structure and increases stiffening. This process is promoted by vascular inflammation and fibrosis.10,11 Activation of p38 MAPK through Ang II is attributed to the increased generation of reactive oxygen species (ROS) causing vascular smooth muscle cell hypertrophy.12,13 In central blood vessels such as the aorta, stiffening of the wall reduces the capacitance properties that usually dampen systolic blood pressure (SBP) elevation. Loss of this function leads to augmentation of the SBP and exaggerates hypertension.14–16 Augmentation of SBP with increased stiffening is a strong predictor of increase of cardiovascular morbidity and mortality17,18; however, whether chronic p38 MAPK directly influences vascular function and reduces blood pressure remains unclear. Therefore, the aim of the present study was to examine whether Ang II-induced hypertension or hypertension-mediated changes in vascular function are dependent on p38 MAPK activation. We hypothesized that p38 MAPK inhibition would ameliorate Ang II-induced hypertension and influence Ang II-mediated changes in the vasculature. To test this hypothesis, we examined the effect of orally available p38 MAPK inhibition through BIRB796, in C57Bl/6 mice.
Materials and methods
Animal care
We obtained 12 week old wild type (WT) mice from an in-house breed at the local animal care facility. The mice were bred on a C57Bl/6J background. Littermates were used as the controls. The animals were housed in Type III Makrolon polycarbonate cages at 45% humidity, 20–22 °C temperature and a 12 h day–night cycle, with free access to water and food. Animal housing and care, and the experiments, took place at the animal care facility of Heinrich-Heine University (Dusseldorf, Germany).
The investigations were conducted according to the US National Institutes of Health (NIH) Guide for Care and Use of Laboratory Animals (NIH, 1996), the NIH publication No. 85–23 revised in 1996. Animal treatment and experiments were conducted with approval of the local ethics committee (O68/08 and G216/08).
Animal treatment
In this study, hypertension was induced in all treatment groups via osmotic minipumps (ALZET Osmotic Pumps, model 1002, DURECT Corporation, Cupertino, CA, USA) with Ang II 1000 ng/min per kg of body weight (BW). Treatment and observation time continued throughout 14 days. Mice were divided into two groups prior to insertion of the minipumps, to either treat with the orally available p38 MAPK inhibitor BIRB796 at a dose of 50 mg/kgBW/d (a generous gift of Boehringer Ingelheim Pharma GmbH & Co. KG, Ingelheim, Germany) or vehicle (the mouse chow, thus oral application) as adopted from previous methods. 19 Treatment started 2 days prior to insertion of the osmotic minipumps and lasted throughout an observation time of 14 days.
For the isolated perfused kidney experiments, mice given a saline infusion only served as the untreated controls.
Treatment groups for chronic p38 MAPK inhibition with BIRB796 were as follows:
Untreated mice (C57B/6) (controls);
Ang II-treated C57B/6 mice (Ang II 1000 ng/kg BW/min) for 14 days, via vehicle; and
Ang II-treated C57B/6 mice (Ang II 1000 ng/kg BW/min) + BIRB796 (50 mg/kg BW/d) for 14 days.
For the ex-vivo inhibition of p38 MAPK (SB203580) in the isolated perfused kidney and thoracic aortic rings experiments, mice were either infused with saline or with Ang II (1000 ng/kg/min) for 14 days.
Isolated perfused mouse kidney
Mice were anesthetized by intraperitoneal injection with ketamine (100 mg/kg) and xylazine (5 mg/kg). Their kidneys were isolated microscopically (Olympus CO11, Olympus Deutschland GmbH, Hamburg, Germany) and perfused with Krebs-Henseleit buffer, according to a method described previously.20,21 Changes in perfusion pressure reflected the changes in vascular resistance of renal vessels immediately after preparation. A bolus injection of 60 mM of potassium chloride (KCl) was delivered to test the viability of the preparation, followed by a stabilization period of 30 min. After the stabilization period, renal vasoconstriction was induced by increasing concentrations of Ang II (Sigma-Aldrich Chemie GmbH, Munich, Germany). Changes in pressor responses were measured in mmHg. To assess vasorelaxation, renal vasoconstriction was induced by norepinephrine at 1 µM (Sigma-Aldrich Chemie GmbH), and then we assessed the concentration-response curves of the vasodilator S-Nitrosoglutathione (GSNO) (Alexis Corp. / Enzo Life Sciences AG, Lausen, Germany). Renal vascular relaxation was calculated as the percentage of contraction in the pre-contracted kidneys, which was set as 100%.
Assessing the acute effects of p38 MAPK inhibition on renal vascular function, the renal pressor response was induced by Ang II in the presence or absence of the p38 MAPK inhibitor SB203580 at 5 µM (Sigma-Aldrich Chemie GmbH). In addition, we assessed the renal vasodilator response to S-Nitrosoglutathione (GSNO) (Alexis Corp. / Enzo Life Sciences AG) in pre-contracted (with 1 µM norepinephrine (Sigma-Aldrich Chemie GmbH)) isolated perfused kidneys, in the presence or absence of SB203580.
Aortic ring myography
We assessed vasorelaxation of the aortic rings from Ang II-treated mice at 14 days with a multi-wire myograph system, as previously described. 22 In short, in the presence of diclofenac (3 µM), the aortic rings were pre-constricted with norepinephrine 1 µM (Sigma-Aldrich Chemie GmbH). We assessed vasodilation by GSNO in the presence or absence of the p38 MAPK inhibitor SB203580 (Sigma-Aldrich Chemie GmbH) at 5 µM. Aortic vasodilation was calculated as the percentage of contraction in the pre-constricted aorta, which was set as 100%.
Immunoblotting for p38 MAPK and phospho-p38 MAPK
Renal cortex tissue was placed into ice-cold 1% Triton lysis buffer (containing a protease inhibitor cocktail (Sigma-Aldrich Chemie GmbH)) and immediately homogenized. Lysates were centrifuged at 20,500 g for 10 min at 4 ºC. We measured protein concentrations using a Bradford assay (BioAssay Systems, Hayward, USA). After dithiothreitol treatment (100 mM) and denaturation (5 min at 95 °C), 30 µg of total protein were loaded onto 10% SDS-PAGE gels and then transferred to nitrocellulose membranes, according to the manufacturer’s instructions (X-Cell Blot Module, Invitrogen / Thermo Fisher Scientific Inc., Oberhausen, Germany). Membranes were treated with blocking buffer (5% bovine serum albumin (BSA) and 0.1% Tween 20 in phosphate buffered saline (PBS)) for 1 h at room temperature, and then incubated either with primary monoclonal rabbit anti-p38 antibody at 1:750 (Cell Signaling Technology, Europe, Leiden, The Netherlands) or primary monoclonal rabbit anti-phospho-p38 antibody at 1:1300 (Cell Signaling Technology), and then mouse anti-β-actin at 1:20,000 (Santa Cruz Biotechnology, Inc., Dallas, USA) overnight. Bound primary antibody was detected with anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody at 1:10000 (Dako Deutschland GmbH, Hamburg, Germany) by 60 min incubation at room temperature. Antibody labeling was visualized by the addition of a chemiluminescence reagent (Lumi-LightPLUS Western Blotting Substrate, Sigma-Aldrich Chemie GmbH). Chemiluminescence was visualized using a FluorChem FC2 Imager (alpha innotec / Protein Simple, San Jose, USA). Immunoblots from each tissue were performed in triplicate.
Quantification of urinary 8-isoprostane concentration
We measured 8-isoprostane as previously described. 23 In short, 24-h urine samples were collected in the metabolic cages at the end of the experimental period. We measured the urinary concentrations of 8-isoprostane using a colorimetric assay kit (Cayman Chemical Company, Ann Arbor, USA). This was normalized to the urinary creatinine concentration.
Quantification of urinary norepinephrine concentration
We collected the 24-h urine samples in metabolic cages, at the end of the experimental period. We measured the norepinephrine concentrations using high performance liquid chromatography (HPLC), as previously described. 24
Histological assessment
At the end of the experimental period, the mice were sacrificed. The mouse aortas were perfused with ice-cold PBS from the left ventricle, embedded in paraffin, and then the abdominal aortas were used for histological assessment.
Ratio of medial area to luminal area in aortas
The aortic rings were embedded in paraffin and cut in 5-µm sections. The slides were stained using a standard PAS (Periodic Acid Schiff) staining protocol. Using the image processing software ImageJ (http://imagej.nih.gov), the lumen and media areas were measured separately, and then the ratio of media to luminal area was calculated.
Quantitative real-time PCR (qPCR)
We used the aorta wall samples to analyze the relative expression of RNA levels for matrix metalloproteinase-1 (MMP-1), matrix metalloproteinase-9 (MMP-9), tissue inhibitor of metalloproteinase 1 (TIMP1), fibronectin and collagen 1. In addition, we analyzed the relative expression levels of the AT1a and AT2 receptor.
After the homogenization of tissue (Tissue Ruptor, QIAGEN GmbH, Hilden, Germany), the total RNA was isolated using a RNA Micro Kit (QIAGEN GmbH, Hilden), according to the manufacturer’s instructions. Quantitative real time PCR was performed with an ABI PRISM 7300 (Thermo Fisher Scientific Inc./Applied Biosystems, Oberhausen, Germany) and a SYBR Green master mix (QIAGEN GmbH, Hilden).
Our study experiments were performed in triplicate. We chose the 18S ribosomal RNA as the endogenous control (a housekeeping gene). The levels of targeted genes were normalized to 18S rRNA expression.
The following primer sequences for mouse cDNA were used:
● MMP-1: ACACTCAAATGGTCCCAAACGAA (forward), GGTGTCACATCAGACCAGACC (reverse); ● MMP-9: CACTGGGCTTAGATCATTCCA (forward), GCTTAGAGCCACGACCATACA (reverse); ● TIMP1: ATCTGGCATCCTCTTGTTGCT (forward), GGTGGTCTCGTTGATTTCTGG (reverse); ● Fibronectin 1: CGAGGTGACAGAGACCACAA (forward), CTGGAGTCAAGCCAGACACA (reverse); ● Collagen 1: CTGGTCCACAAGGTTTCCAAG (forward), AGCTTCCCCATCATCTCCATT (reverse); ● AT1a: GCTTGGTGGTGATCGTCACC (forward), GGGCGAGATTTAGAAGAACG (reverse); and ● AT2: ACCTGCATGAGTGTCGATAGGT (forward), CTGACATCCCGGAAATAAAATG (reverse).
Systolic blood pressure measurement
SBP was measured non-invasively by tail-cuff sphygmomanometer using a BP-98A device (Softron, Tokyo, Japan). Mice were trained for 4 consecutive days prior to the start of the experimental period. Blood pressure was measured on a daily basis. We took 10 consecutive measurements from each mouse, each day at the same time, during the whole experiment. We calculated the mean SBP of each week from the mean blood pressure values of each mouse.
Pharmaceutical drugs used
For the present study, we used the p38 MAPK inhibitors BRIB796 (1-(5-tert-butyl-2-p-tolyl-2H-pyrazol-3-yl)-3-[4-(2-morpholin-4-yl-ethoxy)-naphthalen-1-yl] urea) (a generous gift from Boehringer Ingelheim Pharma GmbH & Co. KG) and SB203580 (4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole, Sigma-Aldrich Chemie GmbH).
These inhibitors predominantly inhibit p38α; however, they also show inhibition of the β, γ and δ isoforms. BIRB796 has a higher specificity for the α isoform, compared to SB203580. Besides inhibition of p38 MAPK, these compounds can also inhibit c-Jun N-terminal kinases (JNKs) to a certain extent. 25 These small-molecule compounds can be applied orally. In this study, BIRB769 was used for oral treatment, whereas SB203580 was used for the ex vivo experiments.
In addition, this study used S-Nitrosoglutathione (GSNO, Alexis Corp. / Enzo Life Sciences AG), angiotensin II (Sigma-Aldrich Chemie GmbH) and norepinephrine (Sigma-Aldrich Chemie GmbH).
Statistics
Data are expressed as the mean ± SEM. Student’s t-test was used to compare the means of two groups with Gaussian distribution. Multiple comparison of more than two groups with Gaussian distribution were analyzed by 1-way analysis of variance (ANOVA), followed by Bonferroni’s multiple comparison post-hoc test. Statistical analyses of data from more than two groups in which the Gaussian distribution was not normal (or could not be assumed) were analyzed by the Kruskal-Wallis Test, followed by Dunn’s multiple comparison post-hoc test. Differences between dose-response curves were analyzed by 2-way ANOVA for repeated measurements, followed by the Bonferroni correction post-hoc test.
Probability levels of P < 0.05 were considered statistically significant. If applicable, a higher level of statistical significance is stated (P < 0.01 or P < 0.001). The number of experiments (n) can refer to the number of mice or the number of individual samples.
Results
Chronic p38 MAPK inhibition attenuates SBP in Ang II-induced hypertension
To evaluate the influence of chronic p38 MAPK inhibition on Ang II-induced hypertension, SBP was measured throughout the observation period. SBP was not statistically different at baseline. Chronic treatment with the p38 MAPK inhibitor BIRB796 attenuated the increase of SBP significantly at Week 1 and Week 2 (Figure 1(a)).
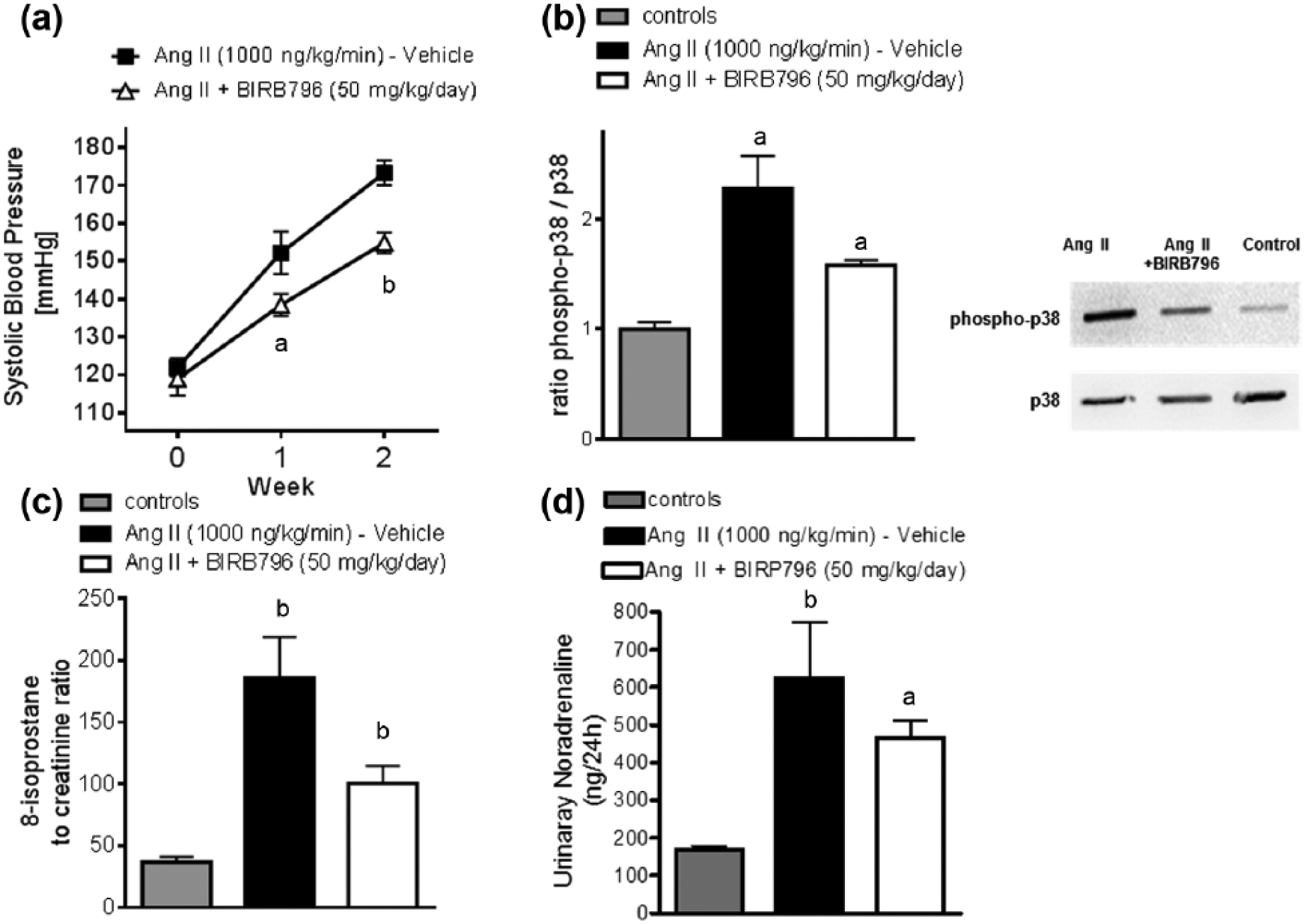
Chronic p38 MAPK inhibition attenuates systolic blood pressure in Ang II-induced hypertension.
In Ang II-induced hypertension, p38 MAPK activation, presented as the ratio of phospho-p38 to total p38 MAPK, was significantly increased compared to controls. Accordingly, chronic treatment with p38 MAPK inhibitor BIRB796 attenuated the increased p38 MAPK activation in Ang II induced hypertension (Figure 1(b)).
This was accompanied by decreased urinary 8-isoprostane excretion in the BIRB796-treated mice, compared to the Ang II-treated controls. Ang II treatment alone significantly increased urinary 8-isoprostane excretion (Figure 1(c)).
Treatment with BIRB796 did not affect urinary norepinephrine excretion, a marker of sympathetic activity (Figure 1(d)).
Chronic p38 MAPK inhibition improves vascular remodeling in Ang II-induced hypertension
Chronic hypertension results in structural changes in aortic wall composition and hypertrophy. 26 Measuring the ratio of media area to luminal area in mice aortas revealed a significantly lower ratio of medial to luminal areas in the BIRB796-treated animals, compared to vehicle-treated animals (0.46 ± 0.02 versus 0.65 ± 0.05; p < 0.01) (Figure 2(a)).

Chronic p38 MAPK inhibition improves vascular remodeling in Ang II-induced hypertension.
We performed qPCR to assess the genes involved in vascular remodeling. Higher expression of MMP-1 and MMP-9 are linked to hypertrophy and stiffening of the aortic vessel wall.27,28 In the BIRB796-treated animals, the relative expression of MMP-1 and MMP-9 was significantly decreased; for MMP1: 0.424 ± 0.180 versus 1.000 ± 0.183 and p < 0.05; for MMP-9: 0.508 ± 0.103 versus 1.000 ± 0.183 and p < 0.05. The stiffening of the aorta is linked to reduced elasticity and increased collagen deposition. These processes are reflected in the reduced matrix turnover and increased collagen and fibronectin expression.28,29 There was no statistical difference in the relative gene expression of TIMP1, fibronectin nor collagen 1 (Figure 2(b-f)).
In the thoracic aortic lysates, p38 MAPK activation presented as the ratio of phospho-p38 to total p38 MAPK was significantly reduced in Ang II-induced hypertensive mice treated with the p38 MAPK inhibitor BIRB796 (Figure 2(g)). Chronic BIRP796 treatment did not affect aortic AT1a and AT2 receptor expression levels (Figure 2(h)).
Improved vascular function by chronic p38 MAPK inhibition in Ang II-induced hypertension
To evaluate whether chronic p38 MAPK inhibition reduces vascular reactivity in Ang II-induced hypertension, the renal pressor response to Ang II was measured ex vivo, in kidneys isolated from mice treated with or without BIRB796 in Ang II-induced hypertension. In BIRB796-treated mice, the pressor response to Ang II significantly decreased, compared to vehicle-treated animals (Figures 3(a) and 3(b)). In contrast, neither the pressor responses to KCl at 60 mM nor to norepinephrine at 1 µM showed any differences (Figure 3(c)). Renal smooth muscle cell-dependent vasorelaxation induced by GSNO was significantly increased in the kidneys of BIRB796-treated mice (Figure 3(b)).

Effects of Ang II and GSNO in the isolated perfused kidney.
Acute inhibition of p38 MAPK improved vascular function in Ang II-dependent hypertension
In order to test for the acute effect of a p38 MAPK inhibition on the vascular function of resistance and conductance vessels, we performed additional experiments in aortic rings and in isolated perfused kidneys of Ang II-treated mice, in the presence or absence of SB203580 (5 µM). In kidneys of Ang II-treated mice, pressor responses to Ang II were significantly higher, compared to controls. Acute p38 MAPK-inhibition significantly attenuated Ang II-induced pressor responses in the kidneys of Ang II-treated mice (Figure 4(a)). Renal vasodilator responses to GSNO were also significantly improved in the presence of SB203580 at 5 µM (Figure 4(b)).
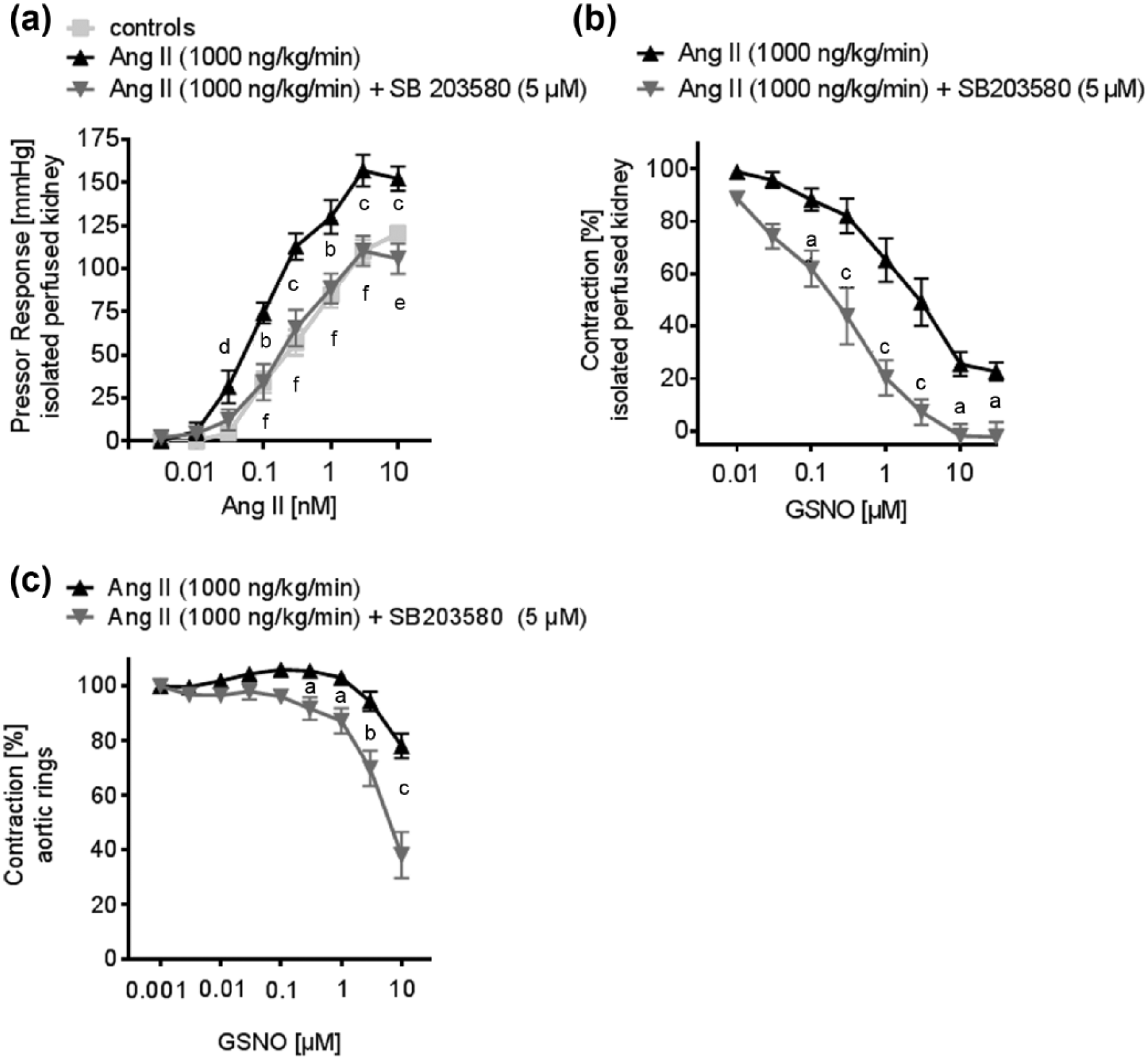
The effect of acute p38 MAPK inhibition on the vascular function of conductance and resistance of vessels was tested in the isolated perfused kidney and thoracic aortic rings.
Discussion
In the present study, we evaluated a novel mechanism of pharmacological intervention in Ang II-dependent hypertension. Our data indicates that p38 MAPK activation plays a crucial role in the development of Ang II-induced hypertension and vascular remodeling. Attenuation of Ang II induced hypertension and improved vascular function by BIRB796 was accompanied by changes in gene expression in aorta vessel wall of markers of vascular remodeling. In addition, the vascular function of the renal resistance arteries was ameliorated by inhibition of p38 MAPK.
In the present study, chronic inhibition of p38 MAPK reduced the SBP in Ang II-dependent hypertension significantly; however, blood pressure levels did not normalize in these mice. The p38 MAPK activation is one of a variety of mechanisms that contribute to Ang II-dependent hypertension. Other mechanisms, such as AT1 receptor heterodimerization and the RhoA/Rho kinase pathway that are activated by Ang II are also very important signaling pathways regulating the extent of Ang II-dependent hypertension.30–32
Recently, Sparks et al. 33 showed that AT1 receptors in renal vascular smooth muscle cells are essential for the development of Ang II-dependent hypertension; however, the underlying mechanism of how AT1 receptors on vascular smooth muscle cells mediate hypertension is not fully understood. In this regard, p38 MAPK seems to be an important player. Herein, the Ang II treatment led to a significant increase in renal p38 MAPK activation and ROS production. This activation was attenuated by chronic treatment with BIRB796 and accompanied by reduced SBP. Previously, we demonstrated that p38 MAPK activation increases pressor responses to Ang II in renal resistance vessels of hypercholesterolemic mice, through ROS-dependent p38 MAPK activation. 4 In addition, p38 MAPK inhibition increased the vasodilator response to nitric oxide (NO) in humans, and decreased the end organ damage in hypertensive rats, through a reduction of ROS abundance.34,35 In this study, the reduced phosphorylation of p38 MAPK through BIRB796 treatment was accompanied by a markedly reduced ROS abundance, as measured by urinary 8-isoprostane, emphasizing the importance of ROS in vascular dysfunction. 23
In this study, chronic inhibition of p38 MAPK not only reduced SBP, but also improved vascular smooth muscle cell-dependent vasodilatation in conductance blood vessels as shown by previous studies. 36 As a significant novelty of the presented study, we could show that chronic inhibition of p38 MAPK attenuated pressor responses to Ang II and improved vasodilatation as a response to GSNO in renal resistance vessels. In contrast to conductance vessels, such as the aorta, resistance vessels play a pivotal role in blood pressure regulation. This finding of the presented study directly links p38 MAPK activation to blood pressure regulation.
Furthermore, the inhibition of p38 MAPK led to a reduction in aortic hypertrophy. These findings are supported by previous studies, which show that p38 MAPK activation is an important mediator of vascular stiffening.13,37 Vascular stiffness is a strong predictor of future cardiovascular events and all-cause mortality. 17
It is well known that p38 MAPK activation mediates hypertrophy of vascular smooth muscle cells in vitro. 38 In our present study, we could show that chronic p38 MAPK inhibition reduced vascular hypertrophy in vivo and it modulated the genes involved in vascular remodeling. Inhibition of p38 MAPK could significantly reduce the expression of aortic MMP1, which is strongly involved in p38 MAPK-dependent vascular remodeling.39,40
In addition, the expression of MMP9, known to be activated by the renin–angiotensin–aldosterone system (RAAS), was also reduced by chronic p38 MAPK inhibition.41,42 MMP9 is a strong inducer of the TGF-β pathway, leading to an increase in collagen deposition, a crucial mechanism for interstitial fibrosis and aortic stiffness.43–46 Based on these results, it is feasible that Ang II-mediated p38 MAPK activation led to vascular stiffening, which itself causes augmentation of SBP. In fact, p38 MAPK activation was reduced in the aortas of BIRP796-treated mice. Differences of AT1a or AT2 receptor abundance cannot be considered as a cause of the attenuated vascular injury in BIRP796-treated mice, as both AT1a and AT2 receptor RNA expression levels did not differ between both groups. Therefore, differences in vascular hypertrophy do not appear to be caused by receptor abundance. In this study, in contrast to previous studies, changes in the relative gene expression of collagen1 and fibronectin, markers of interstitial fibrosis and vascular remodeling, were not changed by our inhibition of p38 MAPK. 47 The relatively short observational period of the study could be an explanation for this finding. Moreover, gene expression of the MMP inhibitor TIMP-1 was also not significantly different in the aortic lysates, in this study.
Besides the described effects on vascular stiffening, there is strong evidence that activation of p38 MAPK influences Ang II-dependent signaling in the vasculature.5,6 Therefore, we examined the acute effect of a p38 MAPK inhibition on vascular function in aortic rings, and in isolated perfused kidneys of Ang II-treated mice, in the presence or absence of SB203580. Control mice were added to these experiments to illustrate the effect of acute p38 MAPK inhibition, compared to subjects of normal blood pressure. Similar to the attenuated renal pressor responses observed in BIRB796-treated hypertensive mice, acute p38 MAPK inhibition reduced renal pressor responses to Ang II to the same extent. Thus, in Ang II-dependent hypertension, the beneficial effect of p38 MAPK inhibition on vascular function is mediated through direct actions on Ang II signaling; and therefore, are at least partially independent from vascular remodeling. Interestingly, p38 MAPK inhibition had no effect on the maximum vasoconstrictive response to KCL nor norepinephrine, in renal resistance vessels. This implied that the observed changes in vasoreactivity were specific to the actions of Ang II. As Ang II-dependent hypertension is dependent on renal AT1 receptor signaling, p38 MAPK signaling in the renal vasculature might be one essential underlying mechanism for the development of Ang II-dependent hypertension. 33
Here we found that acute p38 MAPK inhibition in the isolated perfused kidney and aortic rings obtained from Ang II-treated animals showed improved vasodilation when exposed to GSNO. This might be a result of decreased ROS production. Recently, we and others have shown that p38 MAPK inhibition led to reduced expression of different NADPH oxidase subunits, resulting in reduced ROS abundance.4,34 An increase in ROS abundance contributes to the depletion of NO, a hallmark of endothelial dysfunction. 23 In the presence of increased ROS, the NO scavenging occurs through the formation of peroxynitrite. This mechanism of NO depletion can increase vasoconstriction. Inhibition of p38 MAPK can reduce ROS; and therefore, counteract this mechanism. In addition, p38 MAPK kinase inhibition leads to a reduced MLC(20) phosphorylation in resistance blood vessels. 4 Reduced MLC(20) phosphorylation results in reduced contractility of smooth muscle cells and might potentiate the vasodilatory effect of GSNO in the presence of a p38 inhibitor. Thus, this mechanism might also contribute to the decrease in vasoreactivity and blood pressure through p38 MAPK inhibition, as seen in this study.
In summary, this study elucidated the role of p38 MAPK in Ang II-dependent hypertension. We were able to demonstrate that inhibition of p38-MAPK improves blood pressure and vascular injury through counteracting Ang II-mediated signaling, thus improving vascular remodeling and vascular function. Our findings indicate that p38-MAPK is a potential pharmaceutical target in hypertension.
Footnotes
Acknowledgements
The excellent technical assistance of Blanka Duvnjak, Nicola Kuhr and Christina Schwandt is greatly acknowledged.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Forschungskommission of the Medical Faculty, Heinrich-Heine-University, Düsseldorf, Germany (grant number: 37/2011).
Note
Authors Potthoff and Stamer contributed equally to this work.