
Abstract
Introduction:
Previous studies demonstrated that stimulation of angiotensin subtype 1 receptor (AT1R) led to increased renal generation of prostaglandins E2 (PGE2) and renal inflammation. In turn, PGE2 increases AT1R activity. The conversion of PGE2 to the less active metabolite prostaglandin F2α (PGF2α) via 9-ketoreductase interrupts this feedback loop. The effects of diabetes on the interface between AT1R, PGE2 and PGF2α are not well established. We hypothesized that in diabetes, an aberrant AT1R activity enhances the biosynthesis of PGE2 and impairs the activity of PGE 9-ketoreductase, leading to accumulation of PGE2.
Materials and methods:
Using microdialysis technique, we monitored renal interstitial fluid levels of angiotensin II (Ang II), PGE2 and PGF2α in control and AT1R blocker, valsartan, treated diabetic rats (N=8 each). We utilized the PGF2α to PGE2 ratio as indirect measure of PGE 9-ketoreductase activity.
Results:
Diabetes increased renal interstitial fluid levels of Ang II, PGE2 and PGF2α. PGF2α/PGE2 ratio increased by the third week, but declined by the sixth week of diabetes. Valsartan reduced PGE2 and PGF2α levels and increased Ang II and the conversion of PGE2 to PGF2α.
Conclusion:
Our results suggest that in diabetes, AT1R increases PGE2 generation and reduces conversion of PGE2 to PGF2α with the progression of diabetes.
Introduction
Pathogenesis of the renal complications in diabetes is multifaceted and not completely understood.1–3 Evidence supports a critical role for renin–angiotensin system (RAS) activation in the diabetes-related disturbances in renal microcirculation, inflammation and salt balance.4–10 Inappropriate and uncontrolled activation of the local paracrine RAS sets in motion a series of turbulences in several critical cellular processes which can contribute to the pro-inflammatory changes seen in diabetes. 9 One of these cellular processes affected by hyperglycemia and RAS activation is the COX-derived prostaglandins generation.3,11–13 Among prostaglandins, prostaglandin E2 (PGE2) in particular has a powerful pro-inflammatory effect and has been tightly linked to RAS activation.5,14–25
Previous studies demonstrated that under normal conditions (normal salt and no diabetes), the activity of RAS is minimal, with no effects for angiotensin receptor blockers on basal prostaglandins generation.22,23,25 However, under low salt condition, we demonstrated that AT1R mediates PGE2 generation22,26 and, in turn, PGE2 stimulates renin release and angiotensin II formation.27–29 This crosstalk between AT1R and PGE2 is buffered by the enzyme 9-keto reductase, which increases the conversion of PGE2 to prostaglandins F2α (PGF2α), a prostaglandin metabolite that has an inhibitory effect on RAS.22–26,30,31 The effects of chronic hyperglycemia on the interaction between RAS and prostaglandins E2 and F2α have not been described. We hypothesized that in diabetes, an aberrant AT1R activity enhances the biosynthesis of PGE2 and impairs the activity of the enzyme PGE2 9-ketoreductase, leading to reduction in the inactivation of PGE2 to PGF2α.
Materials and methods
Renal microdialysis technique
To determine the levels of renal interstitial fluid (RIF) angiotensin II (Ang II), PGE2 and PGF2α, we constructed a microdialysis probe as described previously.22,26,32 Each end of a single hollow-fiber dialysis tube (5.0 mm long, 0.1 mm inner diameter) with a 10,000-Da molecular mass cutoff (Hospal, France) was inserted into the manually dilated ends of two 300 mm long (inflow and outflow) hollow polyethylene tubes with 0.12 mm inner diameter and 0.65 mm outer diameter. In vitro recoveries of PGE2 and PGF2α by the dialysis probes were 63% and 60% respectively. Negligible amounts of prostaglandins stick to the polyethylene tubes of the dialysis probes as demonstrated by recovery of 99.5% of PGE2 and 99.7 % PGF2α in the in vitro perfusate.
Animal preparation
Eight four-week-old male Sprague-Dawley rats (Hilltop Lab Animals, Inc., Scottsdale, Pennsylvania) weighing 245–255g were studied. All studies were approved by the University of Virginia Animal Care and Use Committee. For in vivo determinations of PGE2 and PGF2α, the rats were placed under general anesthesia with ketamine (80 mg/kg IM) and xylazine (8 mg/kg IM), and the left kidney was exposed by a left lateral abdominal incision. The microdialysis probes were placed in the cortices of the kidneys while rats were under general anesthesia. Opioid analgesia with buprenorphine (0.01–0.05 mg/kg subcutaneous, administered every 12 h for one day after surgery) was used as our pre-emptive analgesia. No non-steroidal anti-inflammatory drugs (NSAIDs) were used, given the impact of NSAIDs on prostaglandin release. All RIF measurements were made four days after implanting the probes. For collection of RIF, the inflow tubes of the dialysis probes were connected to gas-tight syringes filled with lactated Ringer’s solution and perfused at 3 µl/min. The effluent was collected from the outflow tubes of the dialysis probes for 60-min sample periods in plastic non-heparinized tubes and stored in −80ºC until assayed for PGE2 and PGF2. The animals were rendered diabetic using a single injection of streptozotocin (30mg/kg, Sigma Co., St. Louis, Missouri) 14 days after insertion of the microdialysis tubes. Blood glucose from tail vein was monitored five days after STZ injection (Accu-Check, Boehringer Mannheim Corporation). RIF samples were collected before STZ treatment, and three and six weeks after induction of diabetes. Experiments were started every day at the same time (08:00) to avoid any diurnal variation of the measured substances. RIF samples were collected from conscious rats (N=8) prior to and five hours after receiving the AT1R antagonist valsartan (Novartis Pharma, Basel, Switzerland). Valsartan (10 mg/kg) was administered orally by gavage. Kidneys were examined for fibrosis or inflammatory response surrounding the inserted dialysis probes. 32
Analytical methods
RIF PGE2 and PGF2α levels in dialysate samples were measured by using an enzyme immunoassay kit (Cayman Chemical, Ann Arbor, Michigan). The sensitivity is 50 pg/ml and 40 pg/ml for PGE2 and PGF2α, respectively, and the specificity is 100% for both. The intra-assay and inter-assay cross-reactivity with arachidonic acid or other eicosanoids was <10%. RIF Ang II levels in dialysate samples were measured by using an enzyme immunoassay kit (SPI-BIO, France). The sensitivity is 1 pg/ml and the specificity is 100%. PGF2α to PGE2 ratio is used as an indicator of the 9-keto reductase enzyme activity.22,33–35
Calculations and statistical analysis
All values are expressed as a mean ± standard error of mean. Significance was set at p<0.05 and determined by either unpaired Student’s t-test or one-way analysis of variance (ANOVA) followed by the Tukey test.
Results
Blood glucose at baseline was 86±4.5mg/dl and increased to 362±38 mg/dl five days after receiving STZ (p<0.001). After the induction of diabetes, RIF Ang II recovery levels increased significantly from 0.3±0.02 pg/min to 0.4 ± 0.02 pg/min (p=0.013 vs. baseline) at the third week and to 0.5±0.01 pg/min (p< 0.001 vs. baseline) at the sixth week (Figure 1). There were significant increases in the levels of RIF PGE2, from 17.5±1.4 pg/min to 76.3±14.5 pg/min at three weeks (p< 0.01) and to 133.2±13.1 pg/min (p<0.001 vs. baseline, p<0.01 vs. three weeks of diabetes) at six weeks after the onset of diabetes (Figure 2). Similarly, PGF2α increased from 5.7±0.9 pg/min under normoglycemia to 51.7±3.8 pg/min at three weeks (p< 0.001) and to 60.5±12.4 pg/min at six weeks (p< 0.001 vs. baseline) after the onset of diabetes (Figure 3). RIF PGE2 and PGF2α increased 3- and 9-fold respectively by the third week, and 6- and 11-fold by the sixth week respectively. The ratio of PGF2α/PGE2 ratio (Figure 4) initially increased from 0.3 at baseline to 0.7 by the third week of diabetes. At six weeks of diabetes, the PGF2α/PGE2 ratio was reduced to 0.4, suggesting a decline in activity of the enzyme PGE 9-ketoreductase.

Renal interstitial fluid angiotensin II in conscious rats at baseline and with the development of diabetes (DM), in response to valsartan (n=8; solid line) or placebo treatment (n=8; dashed line).

Renal interstitial fluid prostaglandins E2 (PGE2) in conscious rats at baseline and with the development of diabetes (DM), in response to valsartan (n=8; solid line) or placebo treatment (n=8; dashed line).
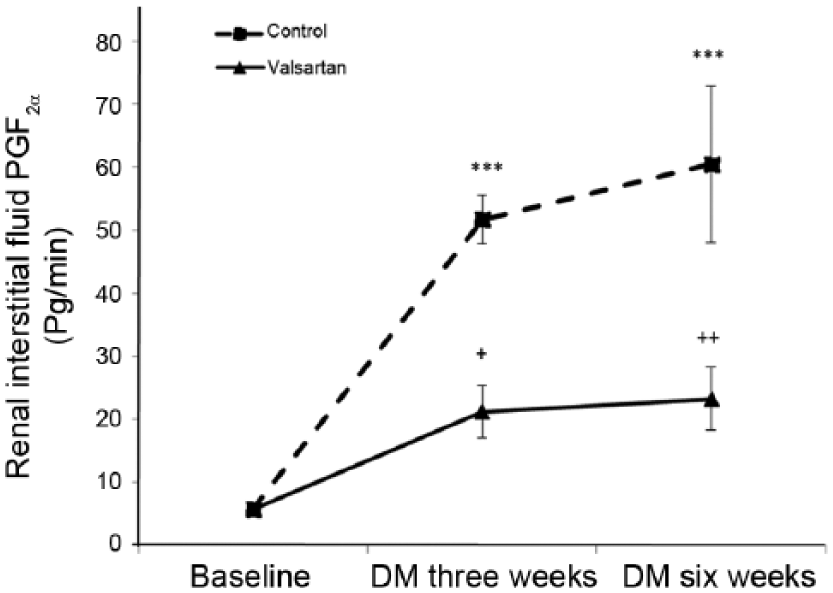
Renal interstitial fluid prostaglandins F2α (PGF2α) in conscious rats at baseline and with the development of diabetes, in response to valsartan (n=8; solid line) or placebo treatment (n=8; dashed line).

Renal interstitial fluid prostaglandins E2:F2 (PGE2:PGF2α) ratio in conscious rats at baseline and with the development of diabetes (DM), in response to valsartan (n=8; solid line) or placebo treatment (n=8; dashed line).
Under normal glycemic conditions, treatment with valsartan did not significantly affect the RIF Ang II, PGE2 and PGF2α levels. The administration of valsartan to the diabetic animals led to a significant increase in the level of Ang II, to 0.5 ± 0.03 pg/min (p< 0.001 vs. untreated three weeks of diabetes) at the third week, and to 0.7±0.05 pg/min (p< 0.01 vs. untreated six weeks of diabetes) at the sixth week. Valsartan treatment also led to a significant decrease in the levels of RIF PGE2, to 35.4±6.8 pg/min at the third week (p< 0.05 vs. untreated three weeks of diabetes) and to 32.6±8.4 pg/min at the sixth week (p< 0.001 vs. untreated six weeks of diabetes) of diabetes (53%, 75% respectively). Similarly, valsartan led to a significant decrease in the level of RIF PGF2α, to 21.1 + 4.1 pg/min at the third week (p< 0.05 vs. untreated three weeks of diabetes) and to 23.2±5.0 at the sixth week (p< 0.01 vs. untreated six weeks of diabetes) of diabetes (59.3%, 61.6% respectively). Valsartan increased the PGF2α/PGE2 ratio from 0.4 to 0.7 at week 6 of diabetes.
Discussion
Accumulating evidence supports a close relationship between the prostaglandin system and the RAS in the kidney. Both systems are also strongly linked to the development of inflammation and the pathogenesis of diabetes complications. However, the interface between prostaglandin and RAS in diabetes remained unclear and mechanistically vague. Prior studies have been hindered by the difficulty in continuous monitoring of early and late paracrine changes in tissues of diabetic animals. The use of the microdialysis technique allowed us to sample in real time and longitudinally in the same, live, and conscious animals the interaction between renal cortical angiotensin II, PGE2 and PGF2α before and after induction of diabetes and in response to AT1R blockade. Here, we demonstrate a role for AT1R in the increased biosynthesis of PGE2 and PGF2α in diabetes and on the conversion of PGE2 to PGF2α.
In alignment with previous reports22,26 under normal conditions, RAS activity is minimal with no effects of AT1R blockade on Ang II, PGE2 or PGF2α levels. Also analogous to our previous report under low salt conditions, we have seen activation of the RAS in early diabetes with a rise in Ang II generation and signaling through AT1R that mediated the increase in prostaglandins generation. Our current study further clarified the effects of chronic activation of RAS on prostaglandins release and the impact of this on the regulation of prostaglandins–RAS crosstalk. The observed increased generation of PGE2 and reduction in conversion rate of PGE2 to PGF2α in diabetes provides a mechanistic explanation of previous reports that suggested an interaction between renin and prostaglandins36–38 and reveals a pathological positive feedback loop with the RAS (increased biosynthesis of prostaglandins and reduced conversion of the RAS-enhancer PGE2 to the RAS inhibitor PGF2α) in diabetes. The increase in the level of PGE2 by the third week of diabetes was associated with a concordant increase in the level of PGF2α in the kidney. Interestingly, while the conversion of PGE2 to PGF2α was initially enhanced by hyperglycemia, this rate of conversion declined by the sixth week of diabetes despite increasing levels of PGE2. This paradoxical early increase in the conversion rate may result from either the increase in PGE2 synthesis or alteration in PGE2 metabolism by 9-ketoreductase or both. A reduction in PGE2 9-ketoreductase activity, however, could only explain the later decline in the rate of conversion of the powerful pro-inflammatory, renin enhancer PGE2 to the less active PGF2α in diabetes, despite increased generation of PGE2. The restoration of this conversion rate with valsartan treatment supports the notion that in diabetes, AT1R has an inhibitory effect on PGE2 9-ketoreductase activity.
In the present study we observed a counter regulatory upregulation of RIF Ang II with valsartan treatment in diabetes. Previous studies suggested that treatment with AT1R antagonists are associated with a rise in plasma Ang II concentration as the result of inhibition of the AT1R-mediated negative feedback on renin release.39–42 Previous studies have also suggested that the resultant increased Ang II stimulates AT2R. 43 Our current study suggests a role for AT1R in reducing the activity of PGE2 9-ketoreductase with the progression of diabetes. We have observed an initial increase in PGE2 9-ketoreductase activity that was followed by decline as diabetes progressed; a potential role for the anti-inflammatory AT2R cannot be excluded and may explain the initial increase in the rate of conversion of PGE2 to PGF2α at three weeks of diabetes (through an intact AT2R). With the progression of diabetes, increase in Ang II and AT1R activity, a failure in AT2R may propagate the reduced activity of PGE2 9-ketoreductase. It is tempting also to speculate that the increased rate of conversion of PGE2 to PGF2α observed with valsartan treatment during the third week of diabetes is mediated through enhancing the activity of the unopposed and functional AT2R. We previously reported a direct role for AT2R in modulating the activity of PGE 9-ketoreductase.22,26 It is possible that the use of AT1R blockade enhances the AT2R function in diabetes. 44 In addition, the observed increase in Ang II levels in diabetes could result from the less opposed inhibitory action of PGF2α on renin release.30,31
Conclusion
The current study suggests that in diabetes, Ang II, through AT1R, plays a critical role in generation and conversion of PGE2 to PGF2α. With progression of diabetes, PGE2 to PGF2α conversion is impaired, leading to further accumulation of the potent pro-inflammatory, renin enhancer PGE2 and the establishment of a vicious cycle between RAS and prostaglandins systems. Given the strong link between RAS, prostaglandins and their downstream signaling pathways mediating angiogenesis, cell adhesion, motility, invasion, morphology, vascular permeability and fibrosis, elucidating the mechanisms involved in the interaction between the arachidonic acid and angiotensin systems may help fine tune future strategies for management of diabetes complications.
Footnotes
Conflict of interest
The authors declare that there is no conflict of interest.
Funding
This work was supported by the National Institutes of Health (grants DK078757 and HL091535 to HMS), the Johns Hopkins Older Americans Independence Center National Institute on Aging (grant P30 AG021334), National Institute on Aging (grants 1R01AG046441 and K23 AG035005) and Nathan Shock in Aging Scholarship Award (PMA)